医学科普
发表者:赵耀忠 人已读
丁寅佳 赵耀忠 汪汇
第二军医大学上海长征医院整形外科
睾丸女性化综合征(Testicular feminization syndrome, TFS),又称雄激素不敏感综合征(Androgen insensitivity syndrome, AIS),是最常见的一种男性假两性畸形。TFS是由于雄激素受体(Androgen receptor,AR)基因突变导致AR结构和功能异常,造成与男性性发育有关的靶组织对雄激素不敏感,从而使雄激素的正常生物学效应全部或部分丧失,进而影响患者向男性化方向的发展。与此同时,由于患者体内雌激素受体正常,而睾酮可经芳香化酶转为雌激素,故使患者青春期后第二性征呈女性表现。TFS属于X连锁隐性遗传病[1],新生男婴TFS的发病率为1/60000~1/20000[2]。
胚胎在性别未分化阶段有一对未分化的性腺,两套生殖管和生殖窦。胚胎的性别发育方向取决于受精卵中的性染色体。若受精卵中包含Y染色体,胚胎将发育成男婴,否则将发育成女婴。这是因为位于Y染色体上的SRY基因,可诱导未分化的性腺发育成睾丸。而胚胎睾丸进而又决定了生殖管的发育,这一过程是由两种激素共同调节完成的,即睾酮(由Leydig细胞产生,促进Wolffian管发育)和Mullerian管抑制物质(由Sertoli细胞产生,抑制Mullerian管发育)。其结果是,两侧的Wolffian管最终成为附睾、输精管、精囊,而Mullerian管退化。但如果缺乏这两种睾丸激素,Wolffian管就会退化,Mullerian管将发育成输卵管、子宫、阴道的上2/3。随着内生殖器的发育,雄激素的持续分泌,将引导外生殖器向男性发展,否则,将向女性演变。未分化阶段的外生殖器主要包括生殖结节、尿生殖褶以及阴唇阴囊隆突。在男性,由于睾酮的衍生物二氢睾酮(DHL)的作用,生殖结节发育形成阴茎头,尿生殖褶变长融合,形成阴茎体和尿道,阴唇阴囊隆突融合形成阴囊。而在女性,外生殖器发育过程外观改变相对不明显,生殖结节形成阴蒂,尿生殖褶形成小阴唇,阴唇阴囊隆突形成大阴唇。此外,雄激素还参与男性第二性征的发育,如骨骼成熟、胡须体毛生长以及精子生成等。
然而,雄激素的所有效应均需要通过AR的参与才能发挥。AR基因位于Xq11-q12[3],包含8个外显子,编码919个氨基酸。其中AR基因的1号外显子编码其转录激活区,2号和3号外显子编码DNA结合区,4号外显子5’端编码铰链区,4号外显子3’端和5~8号外显子共同编码激素结合区[4]。目前已经报道的AR突变位点超过600个,其中绝大部分突变均能引起TFS,仅少部分突变与前列腺癌、乳腺癌的发生相关[5-7]。有70%的AR基因突变是家族遗传性的,由母亲作为携带者传递给子代[8]。
AR基因具有3个特征性的功能域:N末端的转录激活区(NTD)、DNA结合区(DBD)、c末端的激素结合区(LBD)[9]。LBD区域是结合雄激素的关键结构,参与核定位、受体二聚化及蛋白间的相互作用,由4号外显子3’端和5~8号外显子共同编码。AR基因是突变发生率最高的甾体激素受体之一,绝大多数突变发生后可影响受体与配体结合,或者导致受体-雄激素复合物的转录激活潜能丧失。在目前已经发现的600多种突变中,突变方式包括点突变、缺失、插入及剪切点突变等[10],其中以单碱基置换为主且其中一半以上的突变发生在LBD,约20%位于DBD,另有约15%的突变发生在NTD[11]。
AR是核受体超家族成员,位于细胞浆内,是配体依赖性转录因子,与人体内两种主要的雄激素——睾酮和双氢睾酮(DHL)结合,发挥不同的生物学功能。受体-睾酮复合物在胚胎期启动Wolffian管的分化,调控黄体生成素(LH)的分泌和精子发生;受体-DHL复合物的主要作用是促进胚胎期外生殖器官和前列腺的发育,同时参与青春期第二性征的发育[12]。当AR与雄激素结合后发生二聚化,并移位至靶细胞核中,与雄激素下游基因启动子区域或调控区域特异序列结合时,可调节基因的转录,从而引发一系列男性性分化相关的级联反应[13]。
若AR异常,则睾酮与双氢睾酮正常生理作用受到影响即可导致TFS。在胚胎期,TFS患者睾丸Leydig细胞分泌的睾酮由于AR异常,而不能刺激Wolffian管发育形成男性内生殖器。而双氢睾酮对泌尿生殖窦和外生殖器同样不起作用,导致其分化成女性外阴和阴道下段。同时,睾丸Sertoli细胞能分泌正常Mullerian管抑制物质,从而抑制Mullerian管上皮增殖,使其退化,无法发育形成输卵管、子宫、宫颈及阴道上段,最终导致了男性假两性畸形的形成。根据雄激素受体缺陷的程度不同,患者可表现为从完全女性表型,尿道下裂或小阴茎,到正常男性表型仅伴不育或乳腺发育等不同程度男性假两性畸形的广泛表现[2,14,15]。
此外,其他因素如胚胎时期的雄激素水平,1号外显子CAG重复序列的长度,AR与其他共转录因子之间的相互作用,体细胞嵌合现象,5α-还原酶的活性等均可影响TFS患者的最终表型[16,17]。
根据体内雄激素受体的缺陷程度,可将TFS分为完全型和不完全型两类,这两种类型虽然都具有遗传性,但在一个家族中只会产生一种类型的缺陷,临床检查可见睾丸及附睾组织。不完全型患者社会性别常为女性,表现为原发闭经,可见皮肤粗糙,身材高于一般女性,乳房不同程度的发育,男性乳头,无子宫及输卵管,外阴发育存在不同程度的畸形,有少许阴毛,阴蒂肥大,尿道位于阴蒂下方。阴道短浅或呈泌尿生殖窦,睾丸组织一般位于两侧阴唇、腹股沟或腹腔腹股沟内环等处,病理学检查示:曲细精管萎缩,退变严重,管腔闭锁,支持细胞和生殖细胞消失,基底膜增厚,呈玻璃样变,间质细胞呈大片状增生。对外源性雄激素敏感。
而完全型患者皮肤细腻,身材同一般女性,乳房发育良好,女性体态及女性外阴,幼稚型女性生殖器,阴唇往往发育不良,阴蒂不增大,阴道下三分之二发育好,故深度较正常为浅,其顶端为盲端,无子宫,盆腔空虚,阴毛、腋毛稀少,睾丸组织大多位于腹股沟内或腹腔内,病理学检查示:光镜下曲细精管发育良好,偶见精母细胞,未见支持细胞和精子细胞,基底膜无增厚及玻璃样变,间质细胞量少,分散或呈小簇分布。对外源性雄激素不敏感。

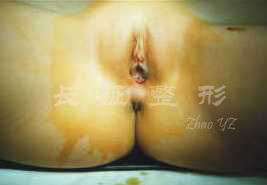
图.1 睾丸女性化完全型患者
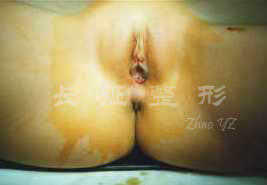
图.2 幼稚型女阴
TFS患者染色体检查核型为46,XY,口腔粘膜检查染色质阴性,荧光小体(Y染色质)阳性。血睾酮水平正常或高于正常男性,雌激素水平相当于卵泡期,阴道脱落细胞检查示雌激素水平相当于月经周期的卵泡期。盆腔充气造影、B超及腹腔镜检查见盆腔空虚,无子宫及卵巢,可有腹腔内睾丸。
对于有TFS家族史的孕妇,应在孕早期或孕中期行羊水或绒毛检查,如胎儿染色体核型为46,XY,孕中期应予B超检查、羊膜腔胎儿造影、胎儿镜或磁共振成像检查观察胎儿外生殖器的发育情况,如检查结果为女性或生殖器发育异常应建议终止妊娠。
关于TFS的治疗常涉及的问题[18]:
1、性腺去留的时机 在切除性腺以避免性腺恶性肿瘤的发生上,目前已获得共识,但对于手术时机仍有争议。Hannema等[19]报道青春期前睾丸的恶变率很小,青春期后约8%,且随着年龄增大睾丸恶变率呈增高趋势;Fallat等[20]报道不完全型患者睾丸恶变率相对较高。睾丸分泌的睾酮可转化为雌激素能促进青春期身高增长及女性第二性征的发育,因此,对于完全型患者,通常建议在青春期第二性征发育后再行性腺切除术(尤其当睾丸位于盆腔内时),而不完全型患者由于睾丸的分泌功能在青春期会出现不同程度的男性化表现,给患者造成心理障碍,睾丸的切除应在青春早期进行比较合适。
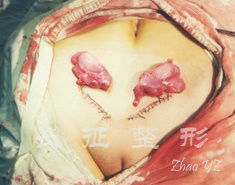
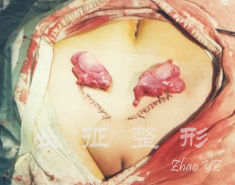
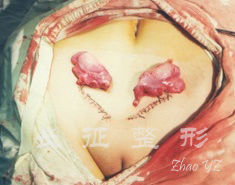
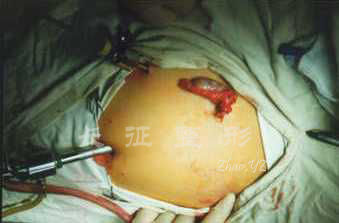
图.3 双侧腹股沟睾丸切除
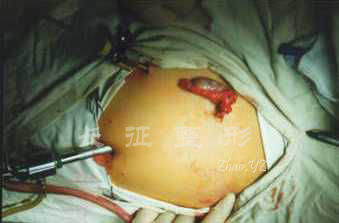
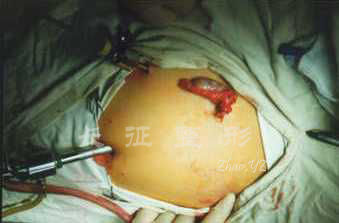
图.4 腹腔镜辅助下腹腔内睾丸切除完成
2、性别取向的指导 人类性别的判定可分为染色体性别、性腺性别、内外生殖器性别、性激素性别、社会性别和心理性别等6个方面。在正常男性或女性,这6个方面性别表现一致。性分化异常者,这6种性别表现可发生多种不一致和冲突。完全型患者大多数其外生殖器表现为发育较好的女性,且常按女性抚养,因此,通常建议按女性性别取向处理。而不完全型患者因外生殖器发展趋向难以预料,而抚养性别的改变会导致将来严重的心理异常,因此,对待该类患者的性别取向的选择尤其需要慎重。
3、性激素的补充 性腺切除后应给予雌激素替代治疗以维持女性第二性征、改善阴道机能及预防骨质疏松,TFS患者因子宫缺如不需同时补充孕激素。有报道称睾丸切除会导致骨密度下降,甚至术后补充常规剂量雌激素,仍有部分患者骨密度下降。雌激素有促进骨骺愈合的作用,补充应待青春发育时从小剂量开始,逐渐加量,以免骨骺过早闭合而导致最终身高偏矮。此外,建议可同时补充钙剂和维生素D。
4、外阴、阴道的整形 完全型患者由于其阴道深度常较正常阴道浅,宽度较正常阴道狭窄,故可行阴道扩张术,以增加阴道的深度和宽度。不完全型患者因存在不同程度的外阴畸形需行相应的整形手术。阴蒂有丰富的血管神经,在性生活中扮演着重要的角色,阴蒂缩小复位术可以保留阴蒂的血管神经,使阴蒂保留良好的功能。若阴道短小可行阴道扩张术,如果失败,也可在婚前半年行阴道成形术,目前国内外主要用回肠、乙状结肠、游离皮瓣、腹膜等代阴道成形术。
5、其它部位的整形 除对患者外阴及阴道进行相应的整形手术外,还可根据患者的具体要求,进行颜面部、胸部等其它部位的整形手术,使患者从外形上与其所选择的性别取向保持协调一致。
参考文献
[6] Heinlein CA, Chang C. Androgen receptor in prostate cancer [J]. Endocr Rev, 2004,25(2):276-308.
[18] 盛少琴, 金杭美. 雄激素不敏感综合征的临床特征及治疗 [J]. 浙江医学, 2011,33(1):92-94.



本文是赵耀忠版权所有,未经授权请勿转载。 本文仅供健康科普使用,不能做为诊断、治疗的依据,请谨慎参阅
发表于:2011-11-09










