




精选内容
-
Krabbe病——表现最像HSP的病之一
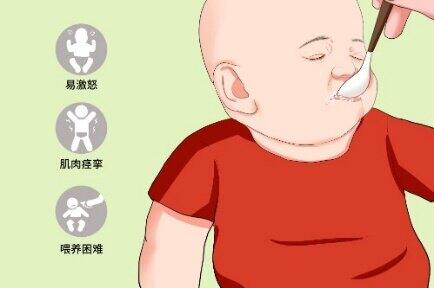
导语:通过前几期,相信大家对遗传性痉挛性截瘫(HSP)有了一定的了解。那么,还有哪些疾病的临床表现和HSP十分相似,又如何鉴别?今天,和大家聊聊Krabbe病。01.什么是Krabbe病?Krabbe病,又称“球样细胞脑白质营养不良(GLD)”或“半乳糖神经酰胺脂贮积病”,该疾病于1916年由丹麦神经学家KnudH.Krabbe首次发现,因此得名。发病率为1/100000至1/200000,是罕见病中的罕见病。02.为什么会得Krabbe病?Krabbe病是呈常染色体隐性遗传的先天性疾病。在某些国家或人种中,该基因突变的人群携带率较高,例如以色列人群携带率为6/1000,其发病率相比其他人群要高。半乳糖脑苷脂酶(GALC)基因,定位在14号染色,基因突变导致GALC酶活性异常从而致病。在神经系统中,GALC酶,是分解代谢某些物质必不可少的重要酶之一,当它的功能或数量发生异常,引起半乳糖脑苷脂不能被顺利降解为神经酰胺和半乳糖,具有神经毒性的鞘氨醇半乳糖苷和半乳糖脑苷脂发生异常蓄积,从而破坏神经轴突周围包裹的髓鞘,导致中枢和周围神经系统广泛脱髓鞘。临床表现为运动、语言、认知、视力和周围神经功能等的异常。03.Krabbe病有哪些症状?按照起病时间,分为早发型(≤6月龄起病)和晚发型(6月龄后起病)。(1)早发型:患儿常有喂养困难、易激惹、阵哭、淡漠、发育迟缓等。该类型发展迅速、预后差,很快出现进行性躯干和四肢肌张力降低、肌阵挛、腱反射亢进和锥体束征阳性,如无治疗,常于2-3岁内死于感染或球麻痹。(2)晚发型:仅占10%,可进一步细分为晚发婴儿型(6月龄-3岁)、幼儿型(3-8岁)、青少年-成人型(>8岁)。起病越晚,症状相对越轻、进展也越慢。成人型,常于30岁前起病,几乎全部病例都表现为慢性进行性加重的行走困难、下肢僵硬(痉挛步态)。部分患者有不对称的肢体无力、锥体束征阳性,也可能伴有认知功能损害和癫痫发作。病程后期,可出现视乳头苍白、球麻痹等。单从临床表型上,极易诊断为痉挛性截瘫;而结合电生理检查结果、肢体的不对称上下运动神经元损害,易被误诊为运动神经元病。但是,绝大多数成人型患者的头颅MRI有异常,从运动皮层经内囊后肢、延伸至脑干的T2相和FLAIR高信号(这能解释患者的下肢痉挛、锥体束征阳性),脑白质也可以呈弥漫性受累、以顶枕部为明显,胼胝体压部多受累,这与肾上腺脑白质营养不良的影像学特点极为相似。04.做什么检查有助于诊断?(1)GALC酶活性检测:外周血白细胞或皮肤成纤维细胞中GALC酶活性测定(低于正常的5%),是帮助诊断的简便且行之有效的证据。酶活性高低与病情严重程度无关。(2)GALC基因检测:通过基因检测发现GALC致病性突变,是诊断金标准。(3)头颅MRI检查:可有脑白质病变、小脑萎缩等表现,是帮助鉴别诊断的重要检查。(4)神经电生理检查:可提示运动和感觉神经传导速度减慢。05.Krabbe病应该如何预防及治疗?首先应避免近亲婚配。患者本人或亲属,在备孕前应进行遗传咨询,优生优育。在某些国家,干血滤纸片法进行酶活性测定,已被用于疾病诊断和新生儿筛查。如有类似症状,应尽早就医!婴儿型,应尽早行造血干细胞移植。晚发型,可使用肌松剂缓解下肢肌张力增高,进行康复等对症治疗。近年,基因治疗、酶替代疗法、小分子伴侣疗等新型治疗方法,分别处于不同的研究阶段。从目前动物模型研究所获得的结果来看,其中一些疗法有望进一步应用于临床,为患者带来新的希望。笔者和团队会进一步关注研究结果,与大家及时分享前沿动态。参考文献:1.SuzukiYandSuzukiKKrabbe‘sgloboidcellleukodystrophy:deficiencyofglactocerebrosidaseinserum,leukocytes,andfibroblasts.Science,1971,171(3966):73-5.2.KomatsuzakiS,ZielonkaM,MountfordWK,etal.Clinicalcharacteristicsof248patientswithKrabbedisease:quantitativenaturalhistorymodelingbasedonpublishedcases.GenetMed,2019,21(10):2208-2215.DOI:10.1038/s41436-019-0480-7.3.ZhaoS,ZhanX,WangY,etal.Large-scalestudyofclinicalandbiochemicalcharacteristicsofChinesepatientsdiagnosedwithKrabbedisease.ClinGenet,2018,93(2):248-254.DOI:10.1111/cge.13071.4.BradburyAM,BagelJH,NguyenD,etal.Krabbediseasesuccessfullytreatedviamonotherapyofintrathecalgenetherapy.JClinInvest,2020,130(9):4906-4920.DOI:10.1172/jci133953.
 田沃土医生的科普号
田沃土医生的科普号 2023年01月14日
2023年01月14日 809
809
 0
0
 5
5
-
脑腱黄瘤病(CTX )简介
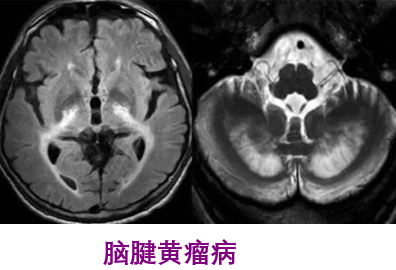
一、概述脑腱黄瘤病(CTX)是一种罕见的常染色体隐性遗传代谢性疾病,是CYP27A1基因突变造成固醇﹣27羟化酶缺陷致病。固醇-27羟化酶缺乏,胆固醇合成胆汁酸受阻,引起不同组织胆固醇代谢障碍。由于体内沉积的胆甾烷醇和胆固醇有神经毒性作用,导致中枢-周围神经系统的广泛损害,出现痴呆、小脑性共济失调、延髓麻痹、进行性下肢疼挛性瘫痪和周围神经病,肌腱黄瘤、青少年白内障和早发的动脉硬化。磁共振检查可见双侧小脑齿状核、小脑白质T2高信号,可累及小脑脚、脑干锥体束及内侧丘系走行区、脑室周围白质、胼胝体和基底核区,随病情进展可出现齿状核T2低信号,可见小脑萎缩。二、临床表现CTX在不同年龄人的身上,症状表现有所不同,婴幼发病的患儿以慢性腹泻症状表现;幼年型患二常出现白内障;少年至青年型患者,则易于多处肌腱出现脂肪堆积的黄瘤;成人型患者可能表现出退行性中枢病变。1.眼睛白内障:约75%患者发生干10岁前,其余25%常发生于40岁之后。其它表征有眼睑黄瘤、视神经萎缩、眼球突出。发生视神经盘苍白者占50%,发生早发性视网膜衰老合并视网膜血管硬化者为30%。青少年时期发生双眼白内障者占90%。2.神经系统表现中枢神经系统在20-30岁间出现锥体系症状者占67%,小脑性共济失调占60%,智能低下(20岁后)占57%,癫痫(约50%)与周围神经病各占24%。小脑体征以及锥体外系有肌张力障碍、类帕金森症状等;神经精神病学症状有幻觉、行为改变、躁动、忧郁等。32%表现为智能发育迟缓,24%出现步态异常。神经系统症状中,锥体系小脑性共济失调占60%,智能低下占57%,癫痫与周围神经病各占24%。3.周围神经病变感觉异常、肢体末端肌肉萎缩及高弓足等。4.黄瘤介于20-30岁间。发生在跟腱、手及手肘伸肌、膝盖及颈部等处;肌腱黄瘤的发生率占56%。跟腱肿物边界清、质硬、动度差、轻度压痛,无搏动感。跟腱MR显示跟腱显著增粗,肌腱中有黄色瘤形成。5.心血管早发的动脉硬化及冠状动脉疾病。6.胃肠道婴幼儿慢性腹泻(最早发生的症状);胆结石(较少见)。7.提早老化早发性白内障、骨质疏松、掉牙、动脉硬化及神经损伤性的痴呆等。8.内分泌甲状腺功能低下。9.影像学表现头颅MRI典型表现有小脑、侧脑室周围白质、基底节、脑干对称性的长T2信号,同时伴有大、小脑萎缩,可见小脑齿状核软化及钙化灶。MRS病灶处乳酸峰及脂质峰增高,NAA峰降低。三、化验检查1实验室检查血浆胆甾烷醇及尿胆汁醇含量异常升高,是诊断CTX的实验室检查最有用的指标。烷醇含量较正常水平高5~10倍,,血浆胆汁醇含量可较正常水平高500~1000倍。脑脊髓液中胆甾烷醇及载体蛋白β上升;皮肤切片培养细胞、血中及肝中的固醇27-羟化酶活性显著偏低。2.影像学检查颅脑MRI见齿状核及小脑,大脑白质的异堂信号。3.基因检查CYP27A1基因缺陷率为99%-100%。四、发病机制本病的可能发病机制是胆酸合成障碍、胆固醇代谢产物胆甾烷醇异常蓄积。健康人体内,固醇27-羟化酶催化胆固醇氧化反应,生成27-羟基胆甾醇,后者再经胆固醇-7α-羟化酶(CYP7A1)催化生成胆酸及鹅去氧胆酸(CDCA),是人体生成胆酸的最主要途径。CTX患者胆固醇27-羟化酶活性不足导致CDCA合成不足,进一步引起胆固醇负反馈调节性升高,通过胆固醇-胆酸经典代谢途径,使胆甾烷醇及胆汁醇含量升高,并在多个系统内大量堆积而引发相应临床症状。五、遗传机制本病的致病基因是CYP27A1,该基因位于2号染色体长臂q33-qter。CYP27A1编码由498个氨基酸组成的固醇27-羟化酶,该基因突变造成固醇27-羟化酶活性大大降低。CYP27A1突变有70余种,其中45%属错义突变,20%无义突变,18%剪接突变,4%缺失突变,2%插入突变。超过50%的突变发生于6号至8号外显子之间,14%位于2号外显子,14%位于4号外显子区域。:绝大多数的错义突变通过影响血红素结合位点及肾上腺铁氧还原蛋白结合位点,破坏固醇27-羟化酶活性;剪接突变导致mRNA快速变性,使翻译表达失败。基因突变类型与临床表型之间并无相关性:有相同症状患者的CYP27A1突变位置及类型可完全不同,在同一家系不同个体之间,临床表现也可有很大差异。六、治疗本病用补充胆酸治疗有效。口服CDCA750mg/d可使血浆胆甾烷醇、尿胆汁醇水平恢复正常,并使患者临床症状明显改善。
 窦肇华医生的科普号
窦肇华医生的科普号 2022年12月12日634
0
0
2022年12月12日634
0
0
-
X- 连锁肾上腺脑白质营养不良(X-ALD)
一、概述X-连锁肾上腺脑白质营养不良(X-ALD)是最常见的过氧化物酶体脂质代谢病,是X染色体上的基因突变所致过氧化物酶功能异常,导致极长链饱和直链脂肪酸不能正常进行代谢,使极长链脂肪酸堆积在血液、肾上腺及脑白质等,引起脑白质脱髓鞘、脊髓退行性变和肾上腺皮质功能减退。本病系ABCD1基因突变导致其编码的蛋白ALDP功能异常。成人X-ALD最主要类型为肾上腺脊髓神经病,临床常表现为痉挛性截瘫。本病为X连锁隐性遗传病,95%以上为男性患者。儿童常在3~10岁发病,约占所有患者的35%。青年以肾上腺脊髓神经病型为主,占所有患者的40%~45%,大多在20~30岁发病。本病预后较差,患者多在出现痴呆、行为障碍等神经相关症状后1~3年内死亡。X连锁肾上腺脑白质营养不良男性发病率为1/21000,女性携带率约为1/14000。二、临床类型与症状X连锁肾上腺脑白质营养不良分7型,不同类型,临床表现不同,主要表现为学习障碍、走路不稳、瘫痪等。本病预后极差,可能会引起一系列并发症,如瘫痪、多个系统功能障碍等疾病,儿童脑型、肾上腺脊髓神经病型患者通常在出现神经症状后3年内死亡。1.儿童脑型10岁以前发病,占35%,临床表现为发病初期患儿注意力不集中、记忆及学习能力下降、行走不稳、视力及听力异常等。部分患者伴有肾上腺皮质功能不全的症状,大部分患儿病情进展迅速,逐渐出现肢体痉挛性瘫痪、共济失调,2~4年内患者完全瘫瘓,呈植物人状态或死亡。2.青少年脑型 发病年龄稍晚,一般11~21岁发病,占4%~7%,症状与儿童脑型类似。3.肾上腺脊髓神经病型21~37岁发病,此型约占40%,平均发病年龄为(28±9)岁,根据是否同时合并脑白质脱髓鞘改变,又可进一步分为2个亚型,单纯肾上腺脊髓神经病型和脑型肾上腺脊髓神经病型。前者仅表现为进行性双下肢痉挛性瘫、性功能丧失等,部分患者有肾上腺皮质功能受损,其病程常超过10年。后者除上述症状外,尚有人格改变、认知功能倒退等脑白质受累症状,病情进展迅速。4.成人脑型 21岁后发病,占1%~2%,脑部呈急性进行性炎症性脱髓鞘改变,不伴肾上腺脊髓神经病变。本型临床表现与儿童型相似,有行为、情绪异常及视力、听力减退等。5.杂合子型 携带致病基因的女性60岁以后可出现本型,极其少见,以周围神经病变为主。6.单纯Addison病 通常在8岁前发病,约占儿童患者的50%,主要表现为肾上腺皮质功能不全的症状,无神经系统病变,大多数患者最终发展为肾上腺脊髓神经病型。7.无症状型 仅有基因异常以及生化改变,临床无明显表现。三、病因X连锁肾上腺脑白质营养不良是由于基因突变所致,基因突变导致大量极长链脂肪酸不能进入过氧化物酶体内进行代谢,堆积在组织与体液中,引发脱髓鞘病变,其好发人群主要是儿童、青年、男性等。1.遗传因素ABCD1基因突变所致。ABCD1位于Xq28,编码ALD蛋白,该蛋白参与将饱和的极长链脂肪酸转运至过氧化物酶体内进行β氧化。ALD蛋白功能异常后,极长链脂肪酸β氧化缺陷,进而在组织与体液中大量蓄积,蓄积到一定程度时,脑白质和肾上腺皮质内的胆固醇酯的脂肪酸构成比发生显著变化,形成胆固醇结晶,使髓鞘的稳定性下降,引发脱髓鞘病变。2.饮食因素 患者饮食长期以极长链饱和直链脂肪酸为主,食用大量高脂食物,属于诱发因素。3.精神因素 长期高度紧张、精神紧绷、过度疲劳等。四、并发症1.多系统功能障碍 X连锁肾上腺脑白质营养不良,尤其是儿童脑型,病情后期临床表现较为复杂,可出现以神经系统病变为主的多个系统功能障碍,如括约肌障碍、深感觉障碍、性功能障碍等,治疗效果一般较差。2.死亡 儿童脑型、肾上腺脊髓神经病型患者发病后3年内,可迅速发展成失明、耳聋及完全瘫痪,最终呈植物状态或死亡,其他分型的患者发展较为缓慢。五、检查1.MRI 颅脑MRI有特征性改变,脑白质呈对称性长T1、长T2信号,可累及胼胝体及脑干,病变由后向前发展,逐一累及枕、顶、颞、额叶,增强扫描病灶的周边区强化,呈“蝴蝶”状。成人X-ALD的MRI特点:(1)肾上腺脊髓神经病患者可见皮质脊髓束、脊髓后索、胼胝体和脑室旁白质信号异常。(2)双侧额叶白质可先受累,对称性的由前向后进展,病灶周围可呈镶边样强化;(3)表现为不对称的局部病灶,容易和肿瘤混淆,部分病例可能与先前头部创伤有关。2.基因检测 脑型X连锁肾上腺脑白质营养不良患者均有ABCD1突变,准确地发现突变类型和突变位点,为携带者筛查、产前诊断和遗传咨询提供可靠证据。3.内分泌检查 成人患者有相当一部分有性功能异常的临床表现及实验室指标异常(如血浆睾酮下降、黄体生成素和卵泡刺激素升高)。肾上腺皮质功能不全者24小时尿17-羟类固醇和17-酮类固醇排出减少,血浆促肾上腺皮质激素升高,促肾上腺皮质激素兴奋试验呈低反应或无反应。六、诊断标准X连锁肾上腺脑白质营养的诊断依靠临床表现、影像学和基因突变分析,头颅MRI检査等,为本病的诊断提供首要线索。有如下4种情况时应考虑本病的可能。1.男性具有痴呆表现、进行性动作发育倒退、视力下降、阅读或语言障碍、书写倒退、协调障碍和其他神经系统表型。2.青年或中年男性有进行性的步态异常,下肢肌力下降或升高,括约肌控制障碍,有或没有肾上腺皮质功能减退等。3.所有具备肾上腺皮质功能减退者,不一定有神经系统症状。4.中年或老年男性表现为进行性截瘫、括约肌控制障碍、下肢感觉障碍等。七、治疗X连锁肾上腺脑白质营养不良的治疗原则包括饮食治疗、皮质激素替代治疗纠正肾上腺皮质功能不全、他汀类降脂药治疗、对症治疗、骨髓移植及基因治疗等治疗方法。X连锁肾上腺脑白质营养不良患者应注重低脂饮食,与此同时,应该合理饮食,注重饮食均衡,多摄入富含维生素和蛋白质的食物,补充体力。儿童注意补充维生素D和钙,促进骨骼生长,年长儿与大人同食,严重植物人状态的患儿可通过鼻饲提供足够营养。八、遗传咨询X连锁肾上腺脑白质营养不良为罕见遗传病,防治关键是产前检查、孕期定期产检、基因检测,早诊断、早治疗,改善患者生存质量。1.早期筛查 产前筛查,定期进行孕检,关注胚胎发育情况。2.预防措施 应科学备孕,进行遗传咨询,优生优育。3.进行定期孕检,确保有无遗传病史。4.按照X连锁隐性遗传方式咨询。早诊断、早治疗是防治关键,因此一旦发现相应症状要及时就诊,同时患者父母、直系亲属应进行基因检测。
窦肇华医生的科普号2022年12月03日291
0
0
-
PSP进行性核上性麻痹
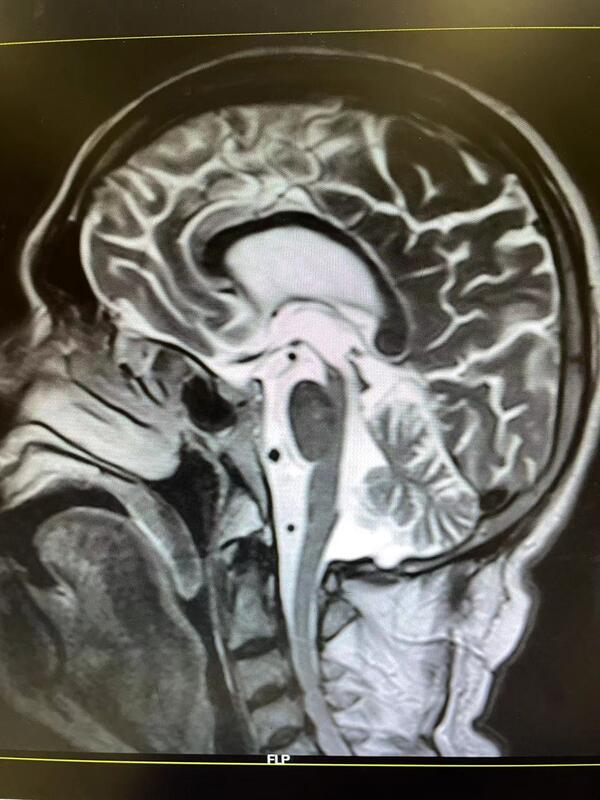
进行性核上性麻痹(Progressivesuperanuclearpalsy,PSP)又称Steele-Rchardson-Olszewski综合征,是一种少见的神经系统变性疾病,以假球麻痹、垂直性核上性眼肌麻痹、锥体外系肌僵直、步态共济失调和轻度痴呆为主要临床特征。PSP的临床表现变异较大,且无特异的实验室检查,极易被误诊。通常开始于中年晚期,进行性核上性麻痹引起肌肉强直,眼球活动不能以及咽部肌肉无力,表现为双眼不能向上转动。由于同时伴发有帕金森病的症状,随着病情的发展,患者可出现严重强直并失去活动能力。本病破坏基底节及脑干,其病因不明,尚无有效的治疗方法。治疗帕金森病的药物有时可减轻其症状。病因可能为变性疾病或病毒感染所致,本病在一定程度上可与Lhermitte综合征、Creutzfeldt-Jakob综合征、Hirano震颤-痴呆综合征以及神经原性色素缺乏综合征的皮质变性重叠,且不能根据其临床表现或解剖变化加以区别,因而目前把这些综合征归为同一类的综合征。临表PSP是一种少见的神经系统变性疾病,以假性延髄性麻痹、垂直性核上性眼肌麻痹、锥体外系肌僵直、步态共济失调和轻度痴呆为主要临床特征。因本病有核上性眼球运动麻痹,故命名为进行性核上性麻痹。本病的临床表现多样,Steele将本病的临床经过分为3期,第Ⅰ期(初期)的临床症状为步行不稳、易跌倒、动作缓慢、视物模糊、语言表达障碍。健忘、易怒、人格改变也可在疾病初期出现。随着病情的发展进入Ⅱ期(中期),会出现眼球运动障碍,主要表现为垂直方向的眼球运动障碍,第Ⅲ期(晚期)则表现为眼球各方面运动不能、躯体僵硬、颈项强直、颈部肌张力障碍明显、不能起立甚至翻身困难。痴呆和语言障碍更加明显,常常出现缄默无动状态。PSP的诊断标准为:中年或中年后起病,进行性非家族性核上性眼肌麻痹至少出现下列5项中的2项:姿势不稳,向后跌倒;假性延髄性麻痹(构音障碍和吞咽困难);少动和强直;额叶综合征(智力迟钝,强握和模仿动作);中轴肌张力异常和强直。多发生于51~60岁男性,平均初诊年龄66.4±7岁,男性多于女性,男女之比约为5:3。隐袭起病,逐渐加重,于发病后2~3年内出现下列症状。1.精神症状逐渐出现性格改变,记忆力减退,智能衰退,很少至严重痴呆。2.核上性眼球运动障碍主要表现为对称性眼球垂直运动障碍。最早为向下注视障碍,继则发生上视运动困难,最后不能水平运动,眼球同定于正中位,瞳孔多缩小,对光反射存在。辐辏反射障碍,呈玩偶眼现象。3.锥体外系症状颈部肌张力障碍为本病的重要症状。出现颈部过伸、仰脸、下颏突出的特殊姿势。头颈部和躯干肌肉明显强硬,四肢较轻,面部表情刻板,皱纹加深,步态不稳,平衡障碍,转身时容易向后方倒倾,但指鼻试验、跟膝胫试验多正常,一般不出现震颤。4.假性球麻痹表现为构音障碍,吞咽困难,下颌反射增强,腱反射增强,可出现病理反射。可有各种非恒定的小脑和锥体束症状和体征。血液、脑脊液和脑电图等检查无异常。气脑造影和CT可见脑室轻度扩大。辅助检查1.脑脊液检查可发现约1/3的患者CSF蛋白含量增高。2.脑电图约1/2的患者脑电图出现非特异性弥漫性异常。3.头颅CT、MRI检查CT对PSP的诊断很有价值,表现有中脑被盖萎缩、中脑水管扩大、脚间池及四叠体池增宽、第三脑室扩大、外侧裂增宽、大脑皮质特别是额叶皮质萎缩、侧脑室扩大等。这些变化会随着病情发展而加重。可见大脑萎缩,MRI检查可显示中脑及脑桥萎缩,伴第三脑室后部扩大,颞叶前部萎缩;T2WI上部分患者可显示壳核低信号。由于MRI可以进行矢状断层,PSP患者萎缩的中脑被盖可以显示得更为淸晰。在PSP患者的磁共振成像正中矢状位上,萎缩的中脑被盖喙在矢状位MRI形似蜂鸟的嘴,因而Kato等首先提出“蜂鸟征”这一征象,其认为蜂鸟征有助于PSP的诊断。尽管中脑被盖的萎缩能为诊断提供重要的依据,但CT、MRI显示PSP患者的外侧裂增宽、额叶萎缩及第三脑室扩大等为PSP的早期影像学表现。中脑被盖的萎缩常较第三脑室、外侧裂增宽及额叶萎缩等出现得晚,所以早期不应过于强调这种改变,以免引起误诊。中脑萎缩和脑干被盖部T2加权像弥散性高信号损害是PSP不同于Alzheimer病和橄榄脑桥小脑萎缩的关键要点。诊断PSP的诊断标准为:中年或中年后起病,向上或向下垂直性核上性眼肌麻痹伴至少有下列5项中的2项:1.对称性运动不能或强直,近端重于远端2.假性球麻痹(构音障碍和吞咽困难)。3.颈部体位异常,尤其颈后仰。4.额叶综合征(智力迟钝,强握和模仿动作)。5.对左旋多巴反应欠佳或无反应Parkinson综合征。一、诊断所需条件(一)纳入条件1.隐匿起病,病程逐渐进展。2.发病年龄≥30岁。3.临床症状:临床症状为并列条件可以同时具有或单独存在。(1)姿势不稳:①病程第1年出现明显的反复跌倒;②1年后出现反复跌倒。(2)病程2年内出现:①垂直性核上性向下或向上扫视缓慢;②凝视麻痹。(3)病程2年后出现:①垂直性核上性向下或向上扫视缓慢;②凝视麻痹。(二)支持条件1.中轴性肌强直或多巴抵抗的帕金森症。2.早期的吞咽困难或构音障碍。3.存在额叶认知功能障碍、冻结步态、非流利性失语或假性球麻痹等无法用排除条件中所列疾病解释的临床表现。4.头颅MRI:正中矢状位T1WIMRI:(1)表现为以中脑萎缩为主的特征性征象:中脑背盖上缘平坦及蜂鸟征;(2)核磁共振帕金森综合征指数(MRPI)=脑桥与中脑的面积比值×小脑中脚/小脑上脚宽度比值>13.55;(3)中脑和脑桥长轴的垂直线比值<0.52或中脑长轴垂直线<9.35mm。5.嗅觉检查和心脏间碘苄胍(MIBG)闪烁显像正常。(三)排除条件1.有其他帕金森综合征病史。2.与多巴胺能药物无关的幻觉和妄想。3.严重不对称性帕金森症。4.采用多巴胺受体阻滞剂或多巴胺耗竭剂治疗,且剂量和时间过程与药物诱导的帕金森综合征一致。5.神经影像学有结构损害的依据(如基底核或脑干梗死、占位性病变等)。6.阿尔茨海默型皮质性痴呆。7.局限性额叶或颞叶萎缩。8.早期出现明显小脑共济失调。9.早期显著的自主神经功能障碍。二、诊断标准(一)临床确诊的PSP-RS必备纳入条件为1、2、3(1)①和(2)②及支持条件4中的两项;无排除条件。(二)很可能的PSP-RS必备纳入条件为1、2、3(1)①和(2)①及支持条件5;无排除条件。(三)很可能的PSP-P必备纳入条件为1、2、3(3)①或②和支持条件1、5;无排除条件。(四)可能的PSP必备纳入条件为1、2、3(1)②或(2)①或(3)①伴有支持条件1、2、3其中一项;无排除条件1-6。鉴别诊断应与早老性痴呆、小脑变性、Parkinson综合征、Alzheimer病及Creutzfeldt-Jacob综合征鉴别。治疗目前对PSP尚无有效治疗方法,神经递质替代疗法是临床治疗的基础。小量抗胆碱能药物可能对控制流涎有帮助;美西麦角可能改善某些患者的吞咽障碍;左旋多巴可能会缓解肌强直,但常因中毒性精神症状而使用药受限;阿米替林小剂量对运动障碍有改善,但大剂量则出现智能及行为障碍等毒副作用,建议用药个体化。预后预后较差,发病后的平均存活期约5.6年(4.8-7.1年),首诊后的平均存活期约为2.7年。常见死亡原因是肺炎包括吸人性肺炎,其次是心血管病如肺栓塞、心肌梗塞、充血性心力衰竭及肾脏感染。
 尚丽医生的科普号
尚丽医生的科普号 2022年10月20日1074
0
0
2022年10月20日1074
0
0
-
没有家族史为什么要做基因检测?
部分患者在经过详细的临床问诊后会发现有特殊的发作规律或诱因,即使未探访到明确家族史,也需进一步完成基因检查,以便评估用药方案及长期预后;比如热敏感性的发作,常见有SCN1A或PCDH19的基因突变,这种突变基因可来源于父母,也可自身新发突变。这种情况是不建议口服钠离子通道阻滞剂类药物的;除此之外,线粒体基因突变所致疾病德巴金是禁忌药物;
冯亚梅医生的科普号 2022年09月27日201
0
1
2022年09月27日201
0
1
-
16p11.2微缺失综合征
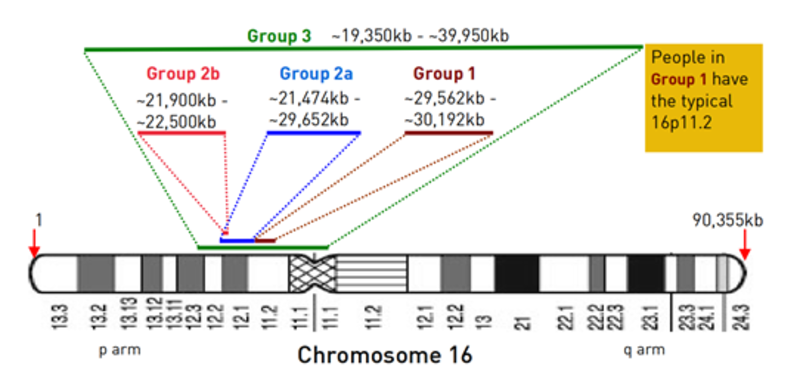
一般来说,16p11.2微缺失者属于以下三种类型之一(见下图)。第一组(Group1):从16号染色体短臂11.2条带开始的约5555万kb的典型微缺失。缺失的遗传信息中含有大约25个已知基因,其中只有一部分的功能已明确。16p11.2带碱基对只有一个拷贝。第二组(Group2):大小不一的缺失,不与典型的微缺失重叠,是从它侧面的区域,靠近16号染色体短臂的未端。第三组(Group3):较大的缺失包括典型的微缺失。16p11.2遗传物质的丢失或重复是最常见的结构染色体疾病之一。典型的16p11.2微缺失因为缺失片段太小,无法用核型方法检测到,需要由微阵列(CGH或CMA)来进行诊断。最常见的症状有:1.语言发育迟缓;2.部分表现为发育迟缓和学习障碍;3.发育迟缓,相较于运动方面,更多的会影响思考和语言方面的能力;4.自闭症或自闭症谱系障碍易感性增加;5.非常微小的面部或身体异常;6.婴儿常表现为肌张力低下:7.超重倾向;8.少数有癫痛发作;9.每一位16p11.2微缺失综合征患者,即便是同一个家庭的成员,其症状和受影响程度也各不相同;10.16p11.2微缺失综合征患者通常无重大出生缺陷;11.有些人完全不受16p11.2微缺失的影响。没有学习、语言或生长困难;12.携带16p11.2微缺失的人(不论男女),会将其遗传给后代,后代的临床表征与父母会有差异。是否有16q11.2微缺失携带者为健康人,没有重大健康问题或出生缺陷,并正常生长发育?是有的。当14个携带16q11.2微缺失个体的父母进行染色体检查时,有三位完全正常和健康的父母(两位母亲,一位父亲)与其受影响的孩子携带有相同的16p11.2微缺失。在另一项研究中,大约每19000个普通人中就有两个携带有16p11.2微缺失。语言发育迟缓 语言发育迟缓是常见的,这可能是发育迟缓的第一个征兆。这种延迟尤其影响表达性语言,而理解力相对保留。语言发育的延迟可以不依赖于任何学习困难而独立存在。在26位携带者中,20位有明显语言发育迟缓。说第一个可以辨认的词的年龄从12个月到7岁不等,说话可能会出现得更晚。发育迟缓 2/3的患儿有发育迟缓的症状。每个人的延迟表现和严重程度是有差异的,在语言、粗大运动和精细运动方面都会有不同程度的影响。一些家庭报告说孩子在某些领域能力学习超前,在其他领域则有延迟。一旦延迟得到确认,尽早开始治疗是有帮助的。活动能力 活动能力通常比思考能力受到的影响要小。婴幼儿在坐姿、爬行和行走速度可能较慢,但到目前为止,每一位微缺失者都能够走路,通常只比一般发育中的儿童稍晚一点。精细运动 精细运动发育迟缓,如婴儿很难抓住和玩玩具,也很难掌握钳子的握法。但表现因人而异,一些儿童的精细运动没有明显影响,而其他儿童受到的影响比活动能力更大一些。自理能力 儿童能够达到的自理能力水平个体差异较大。精细运动发育迟缓和学习困难较大的儿童,需要更多的帮助;精细运动发育较好、学习困难程度较轻的儿童,在自理方面可以变得非常独立。学习障碍 通常16p11.2微缺失的患儿,学习能力范围从正常到轻微的延迟。有些完全没有学习障碍,少数在特定学习领域有学习天赋;另一些有特定学习障碍。多数的学习障碍可以被描述为“缓慢正常”或轻微的,智商在60-79之间;还有另一些人在学习方面面临更大的挑战,难度从中度到重度不等,需要更多的支持。对自闭症或自闭症谱系障碍易感性的影响 在自闭症或自闭症谱系障碍(如阿斯伯格障碍)的儿童和成人中,典型的16p11.2微缺失比在普通人群中更常见。并不是每个微缺失的人都有自闭症,大概是1/3到1/5的比例。男孩更容易受到影响,且受影响程度较大。面部或身体特征有轻微异常16p11.2微缺失的儿童和成人看起来并不是特别相像,也没有可识别的相似的面部或身体特征。大部分病例有较为相似特征,即异常大的头部;与之相反,16p11.2微重复的儿童头部较小。超重倾向 16p11.2微缺失的儿童和成人中,几乎有一半人有超重和肥胖的倾向。在婴儿时期,超重会伴随一段时间,尽管喂养得很好,但体重增加很困难。在儿童时期,体重差异较大,有些孩子又瘦又小,但最迟在成年期会出现肥胖。2岁以下,不受影响;儿童期可能出现超重或肥胖;青春期和成年期出现肥胖。有一对16p11.2微缺失的双胞胎,在28岁时,他(她)们的身高和他(她)们未受影响的哥哥差不多,但是比哥哥更重,有84-88kg,他(她)们的哥哥只有71kg。一个可能的致病基因是SH2B1,其他基因可能也起了作用。在一项研究中,丢失一份SH2B1拷贝的儿童,在孩童时期体重迅速增加,他(她)们的体重增加大部分是由脂肪引起的。他们有明显过量饮食倾向,以及升高的胰岛素水平,并伴有明显的胰岛素抵抗。总的来说,生长发育似乎没有受到影响。少数有癫痫发作 一般来说,16p11.2微缺失的人身体健康。目前还不确定癞痫发作是否比其他一般发育中的儿童更常见。数据表明,四分之一患有癡痫或有过癡病,药物可以有效控制发作。在儿童期,癫痫常会得到緩解或变得更温和。癡痫通常发生在婴儿期和青春期之间。在一项研究中,16位携带16p11.2微缺失患儿,有6位有行为问题,如注意缺陷多动障碍[ADHD],也称多动症。而另一项研究表明,在21位16p11.2缺失患者中,未发现过度活动的高发生率。根据沈雅之资料编辑
 窦肇华医生的科普号2022年09月23日1807
0
3
窦肇华医生的科普号2022年09月23日1807
0
3
-
马歇尔-史密斯综合征/罕见病
马歇尔-史密斯综合征是一种先天性疾病,由于基因突变所引起的,目前尚且无法治愈。导致马歇尔综合征的基因被称为NFIX。患儿通常会出现眼球突出、牙齿向前伸展等头面部和肢体缺陷。同时,还会伴有智力、听力、语言和运动障碍以及呼吸困难等症状。
付朝杰医生的科普号 2022年09月17日997
0
0
2022年09月17日997
0
0
-
22q13缺失综合征
一、 概述22q13.缺失综合征,也称Phelan-McDermid综合征,是指22号染色体长臂末端微小缺失或重排导致的一种综合征。该片段在染色体末端附近,发生在指定的q13位置。22q13缺失综合征的特征变化很大,涉及身体的许多部位。特征性体征和症状包括发育迟缓,中度至深度智力障碍,肌张力降低(张力减退)以及言语缺失或延迟,自闭症或类似自闭症的行为,这些行为会影响沟通和社交互动,例如眼神接触不良,触摸敏感、攻击性行为及咀嚼衣物等非食品,还有癫痫发作。该综合征的个体倾向于对疼痛的敏感性降低。许多人的出汗能力也降低,这可能导致更大的过热和脱水风险。有些人有周期性呕吐、恶心和胃食道反流。已知至少500例22q13缺失综合征。二、临床表现22q13缺失综合征的人通常具有以下主要特征。1.头颈部 巨头、长头(57%);面部两侧不对称、弓形眉、上颌前突、小下颌(62%);上睑下垂(57%)、内呲赘皮;外展耳、外耳发育不良(65%)、听力障碍。2.生长发育 身材高大、发育速度接近正常(95%)、新生儿肌张力低下与喂养困难。 3.神经系统 中枢神经系统:运动发育迟缓、语言发育迟缓(99%)、中度至重度智力发育迟缓、全身肌无力(97%)、癫痫、体温调节能力差(51%);周围神经系统:疼痛耐受性增高(86%)、新生儿发射减弱及反射异常;精神行为异常:咀嚼行为异常(70%)、磨牙、吐舌、孤独谱系障碍倾向、社交能力差、攻击行为。4.四肢 手掌大、肉质手(68%)。 5.皮肤与趾甲 易发热、少汗、趾甲发育不良(87%)。三、发病原因22q13缺失综合征是由缺失引起的靠近22号染色体的长(q)臂末端。22q13缺失综合征的体征和症状可能与该区域多个基因的丢失有关。缺失的大小因受影响的个体而异。一环染色体22也可引起22q13缺失综合征。环状染色体是一种环状结构,当染色体在两个地方断裂,染色体的尖端丢失,断裂的末端融合在一起时发生。具有环22号染色体的人在其部分或全部细胞中具有该异常染色体的一个拷贝。当环形染色体22形成时,22号染色体长(q)臂末端附近的几个关键基因会丢失。如果其中一个染色体断裂点位于22q13位置,则会出现22号环染色体的人具有与简单缺失的那些相似的体征和症状。导致22q13缺失综合征特征的所有基因中,SHANK3很可能是负责许多综合征的特征性体征(如发育迟缓,智力残疾和言语障碍)。缺失区域中的其他基因可能有助于22q13.3缺失综合征的不同特征。四、遗传模式22q13缺失综合征的大多数病例不是遗传的。该片段最常发生在生殖细胞(卵子或精子)形成期间,或胎儿早期发育过程中的随机事件。患者通常没有家族疾病的病史。22q13缺失综合征患者以常染色体显性遗传方式遗传给后代。大约15%至20%的22q13缺失综合征患者父母健康,但亲本是染色体平衡易位携带者,其中来自一条染色体的片段与另一条染色体的片段进行交易,但没有获得或丢失遗传物质。染色体平衡易位传递给下一代时,它们会变得不平衡。继承不平衡易位的儿童可以进行具有额外或缺失遗传物质的染色体重排。
窦肇华医生的科普号2022年09月12日883
0
0
-
祖祖辈辈没有患遗传病的,为啥不能排除遗传性疾病?
经常听家长朋友说:“我们家祖祖辈辈没有得遗传病的,孩子不可能是遗传病。”有些将信将疑,但还是遵医嘱进行了遗传性疾病相关检查。有些根本不信,也不进行相关检查。不幸的是,有些运气不佳的家庭又迎来了第二个患相同疾病的孩子。令人感觉非常遗憾和无奈。群众口中的遗传病一般是指这个病是从祖辈传过来的(从父辈或者祖父辈)。医学上的遗传病是指遗传物质发生改变所导致的疾病。人类的遗传物质是DNA,绝大部分存在于细胞核内的染色体上(核DNA),少部分在线粒体内(线粒体DNA)。我们的遗传物质都来自于上一代(核DNA的一半来自父亲,一半来自母亲;线粒体DNA都来自母亲)。胚胎发育早期,受精卵形成后,精子和卵子携带的遗传物质组合在一起,为下一代提供一整套完整的遗传信息。在胚胎发育期,受精卵细胞的复制分裂伴随着遗传物质(DNA)的复制和再分配(到各个细胞内)。过程中受各种因素的影响如药物、病毒感染、射线辐射、噪音等等,遗传物质在复制分离过程中如果出现了差错,就产生了各种各样的染色体畸形、基因变异(所以,孕期保健避免各种有害因素是非常重要的)。所以,遗传性疾病可以是在某个个体新产生的。因此,广义的遗传性疾病应该有三层意思:1,是遗传物质改变导致的疾病;2,遗传物质的改变可以来自于上一代,也可以是新产生的;3,新产生的染色体异常或基因变异可以传递给下一代。所以,祖祖辈辈没有患遗传性疾病,还是不能排除患遗传性疾病的可能。尊重临床医师的判断,进行相应的检查才是对孩子、对家庭负责的表现。
常杏芝医生的科普号 2022年09月01日795
0
4
2022年09月01日795
0
4
-
现在还没有药物治疗,确诊的意义在哪里?
转自:顾大夫视频号-1、由患者或者家庭成员在充分知情的基础上做出适合的决策。2、知情选择:医患需要尽量调节到一个频道进行交流,这是很难的,时间、知识落差,患者情况千差万别,自己可能描述不清。3、诊断是针对性治疗的前提,如果不知道是什么病,怎样来进一步谈具体治疗的方法?4、现在没有药物,具体指什么?5、罕见病:病种很多,不同的疾病、不同的病程阶段、不同的患者家庭背景情况,需要综合考虑。6、确诊的意义在哪里?(1)减少盲目就医,减少浪费时间和资源;(2)与专业医生和研究者团队、以及同一种疾病的病友群体建立精准连接;(3)针对性地了解这个疾病目前的研究进展,可以更快更准确地获取资讯,提高临床试验的参与度,患者需要参与研究。(4)遗传病:阻止致病基因的传递,明确家族的病因,对于整个家族的后代非常重要。

 顾卫红医生的科普号
顾卫红医生的科普号 2022年09月01日978
0
2
2022年09月01日978
0
2
相关科普号
冯亚梅医生的科普号
冯亚梅 主治医师
上海德济医院
癫痫中心
959粉丝18.6万阅读

袁云医生的科普号
袁云 主任医师
北京大学第一医院
神经内科
9963粉丝152.3万阅读

张伟医生的科普号
张伟 副主任医师
北京博爱医院
神经康复科
51粉丝13万阅读
-
 推荐热度5.0孙一忞 副主任医师复旦大学附属华山医院 神经内科
推荐热度5.0孙一忞 副主任医师复旦大学附属华山医院 神经内科帕金森 9票
共济失调 9票
锥体外系疾病 6票
擅长:运动障碍、神经系统遗传病、神经系统变性病的诊治,如帕金森病、帕金森综合症、肌张力障碍、痉挛性截瘫、舞蹈症、抽动症等。神经系统单基因病和多基因病的遗传咨询。 -

 推荐热度5.0朱国行 主任医师复旦大学附属华山医院 神经内科
推荐热度5.0朱国行 主任医师复旦大学附属华山医院 神经内科癫痫 141票
睡眠障碍 11票
头痛 7票
擅长:癫痫、头痛、睡眠障碍、脑炎(自身免疫性脑炎)、神经痛及脑血管病等神经内科常见病和疑难杂症的诊治,以及脑电图的判读分析 -

 推荐热度4.9许保磊 副主任医师宣武医院 神经内科
推荐热度4.9许保磊 副主任医师宣武医院 神经内科帕金森 48票
锥体外系疾病 22票
痉挛性斜颈 2票
擅长:1、震颤,帕金森病,继发性帕金森综合症,帕金森叠加综合症(进行性核上性麻痹,多系统萎缩,皮质基底节变性等),肌张力障碍 帕金森病风险评估,DBS手术咨询 2、痴呆(帕金森病痴呆,路易体痴呆,额颞叶痴呆,血管性痴呆,阿尔茨海默病等) 3、脑血管病,脑梗死,脑出血,脑血管狭窄,头晕,头痛等 尤其擅长帕金森及相关疾病诊断及治疗