



科室介绍 查看全部
科室医生 查看全部
-
骨科

梁加利
副教授
骨科主任
骨科吴建斌
主任医师
3.6
骨科
何祖胜
副主任医师
3.4
骨科
刘照华
副主任医师
3.4
骨科金文涛
主治医师
3.4
骨科邝冠明
副主任医师
3.4
骨科费志强
主治医师
3.4
骨科陶超雄
主治医师
3.4
骨科高迪
主治医师
3.4
骨科娄楠
主治医师
3.4
-
骨科
郭海华
主治医师
3.4
骨科
周亚鹏
主治医师
3.4
骨科王飞
主治医师
3.4
骨科王磊
主治医师
3.4
骨科尹世杰
主治医师
3.4
骨科李翔
主治医师
3.4
小儿骨科
董忠信
主治医师
3.2
骨科陆瓞骥
教授
3.3
骨科张文智
教授
3.3
骨科曲广运
教授
3.3
-
骨科
叶永玉
副教授
3.3
骨科丘伟鹏
副教授
3.3
骨科杜启峻
3.3
骨科忻振凯
3.3
骨科黄德民
3.3
骨科钟佳权
医师
3.3
骨科余志才
医师
3.3
骨科高庆鹏
医师
3.3
骨科刘德福
医师
3.3
骨科冯松
医师
3.3
-
骨科
林云志
医师
3.3
骨科吴天顺
医师
3.3
科普·直播义诊专区 查看全部
- 精选 干骺端软骨发育不良,Schmid型
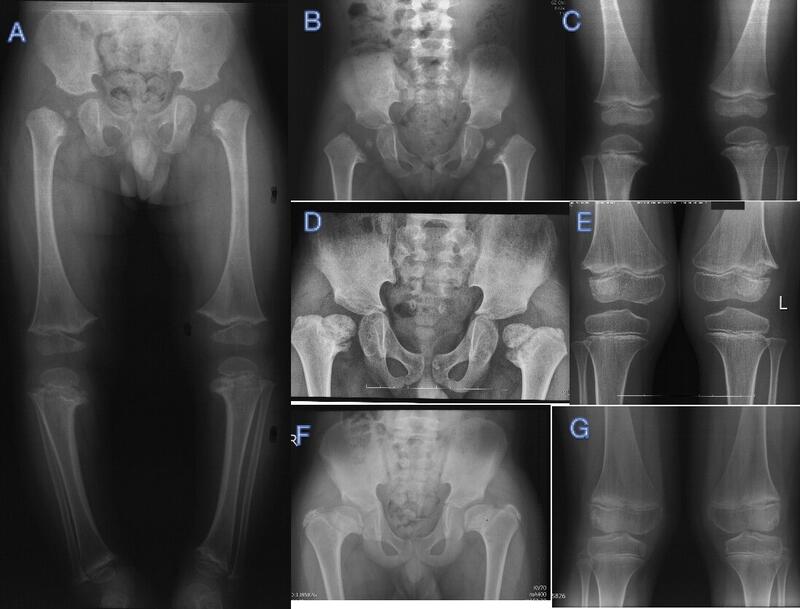
临床表现干骺端软骨发育不良,Schmid型(MCDS)是最常见的、最轻型的MCD类型,与最严重的Jansen型同为常染色体显性(AD)遗传。MCDS以轻度身材矮小(短肢型)、膝内翻(O型腿)、髋内翻为主的骨科表现,常在开始走路时(1-2岁)因O型腿和(或)摇摆步态就诊。随着年龄增大,O型腿有自行或非手术治疗改善可能,但髋内翻常需截骨手术治疗。上肢长骨干骺端可出现下肢同样改变,但常无需特别处理,脊柱改变少见,偶见扁平椎和终板不平整。鉴别诊断佝偻病:两者发病年龄和X线表现高度重叠,临床上MCDS常被误诊为佝偻病,但营养不良性佝偻病和低磷性佝偻病在血生化学检测都可见到异常。其他类型干骺端软骨发育不良:有其他系统受累表现,Jansen型侏儒最严重,合并眼距增宽、眼球突出表现,由3号染色体上的甲状旁腺激素PTH/PTHrP受体基因突变导致,类似甲状旁腺激素亢进,有严重的高钙低磷血症。其他型MCD为AR遗传,McKusick型也称软骨毛发发育不良,芬兰人多见,合并毛发稀疏、贫血和免疫缺陷等血液系统异常。Shwachman-Bodian-Diamond综合征可有胰腺外分泌不足和中性粒细胞减少。另外基因检测可明确各型MCD的突变位点。脊柱干骺端发育不良:脊柱X线可见异常表现明显。骨科治疗因为MCDS无骨科以外系统受累,骨骺正常,骨成熟后关节功能受影响小,临床预后较好。骨科以治疗髋内翻和膝内翻为主,当出现摇摆步态(鸭步)、疼痛、髋内翻进行性加重时常需手术治疗,支具、牵引、物理治疗等保守治疗多无效;髋内翻多采用股骨近端外翻截骨手术治疗,有多种术式,其目的都在于改善力线,恢复髋外展肌力臂,刺激股骨头和颈骨缺损愈合;MCDS膝内翻有一定自愈能力,支具可用于轻症患者治疗,生长期儿童严重的膝内翻畸形可通过骨骺阻滞术治疗。严重髋内翻多伴随着代偿性膝外翻,处理髋内翻时要同时处理膝外翻为宜。
 董忠信 主治医师 香港大学深圳医院 骨科50人已读
董忠信 主治医师 香港大学深圳医院 骨科50人已读 - 精选 假性软骨发育不全、多发骨骺发育不良、软骨发育不全
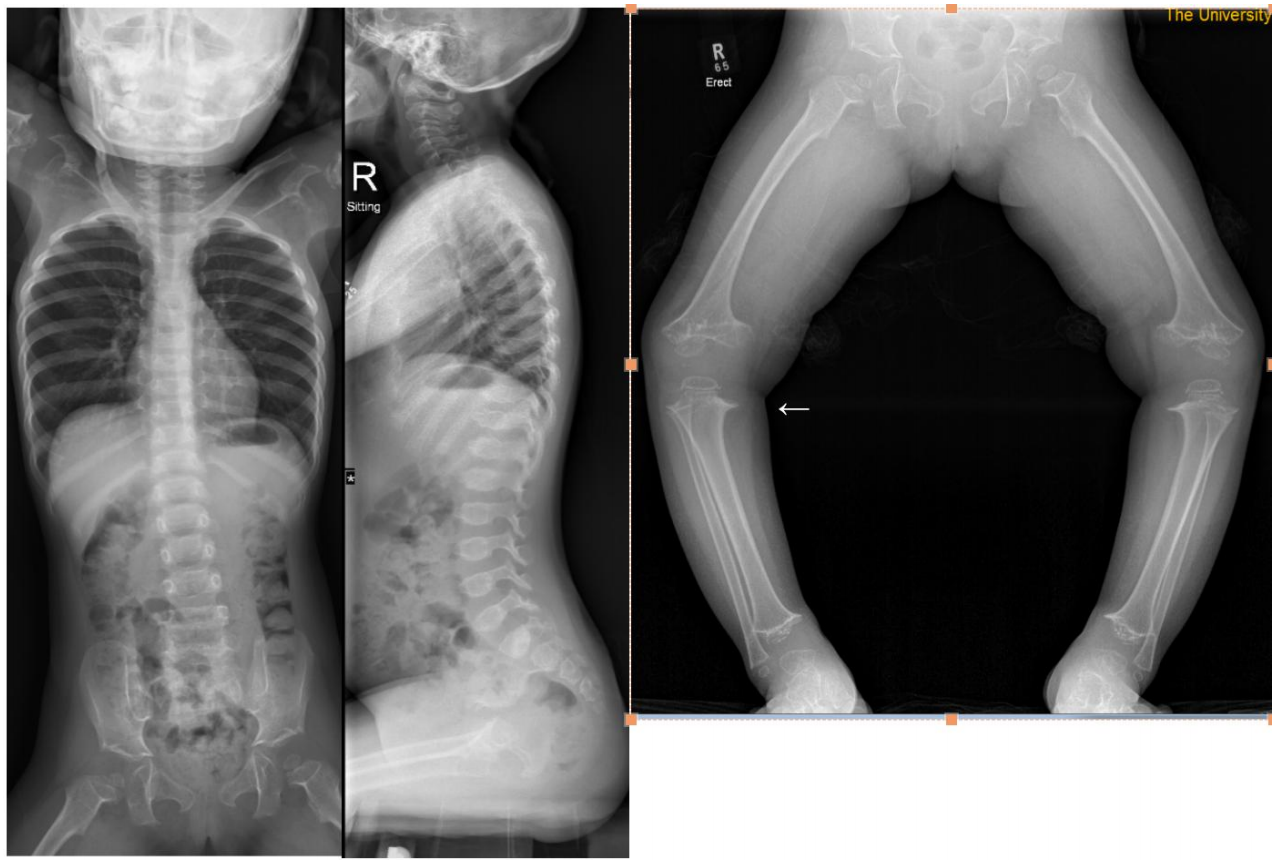
假性软骨发育不全(Pseudoachondroplasia,PSACH,OMIM:177170)是一种相对常见的常染色体显性骨骼发育异常疾病,主要由软骨低聚物基质蛋白(COMP)基因突变导致,也有COL9A3基因突变报道;其特征为不成比例的矮小、关节松弛、早发型骨关节病和脊柱骨骺干骺端发育不全。1959年最早由Maroteaux和Lamy报道,国外群体中PSACH发病率约为1/30000,而我国尚无明确的PSACH患病率调查。临床表现•颅面部正常,无畸形特征;智力正常;•出生时身长正常,20月龄后出现侏儒症状;中度、严重的短肢型矮小,生长速度下降到低于标准生长曲线约2岁;成年后平均身高男性为120厘米,女性为116厘米。•轻微的指过短;•韧带松弛和关节过度伸展,特别是手、膝盖和脚踝;开始走路时出现特征的蹒跚步态;•一些人报告有轻度肌病;•肘部和臀部的伸展受限;•下肢内翻为主,外翻或风吹畸形也可见到;•儿童时期的关节痛,特别是下肢的大关节;上肢和脊柱的骨关节炎可能发生在成年早期。退行性关节疾病是进行性的,约50%的PSACH最终需要髋关节置换手术。•轻度脊柱侧凸:可在儿童时期发现,并可持续到成年。齿状骨发育不全并不常见,但有时也会发生,可能导致颈椎不稳,出现一系列神经系统并发症;•腰椎前凸(约占50%的患者);没有软骨发育不全明显。影像学特征•长骨骺和干骺端同时受累,延迟骨化伴长骨骺端和干骺端不规则;•股骨头骨骺小,短股骨颈,干骺端不规则,呈喇叭形;骨盆小,髋臼边缘不规则,易硬化;•显著的指过短;短的掌骨和指骨显示小的或圆锥形的骺端和不规则的干骺端;小而不规则的腕骨;骨龄延迟;•脊柱可有轻度胸腰区后凸,腰椎前凸,一些患者可见到脊柱侧弯;侧位X线上椎体的中间突或舌突,椎体后缘无凹陷;椎体的这种独特外观随年龄的增长而趋于正常,这强调在儿童时期进行用于诊断的x线片的重要性;诊断假性软骨发育不全的诊断是通过遗传史、具有上述典型的临床和影像学特征;青春期前患儿的X线片,至少需要双下肢全长片、脊柱全长正侧位和手腕正位。但如果上述临床特征不典型,通过基因检测到致病基因突变可以明确诊断。截止2019年4月,HGMD已收录107种COMP致病性基因突变与假性软骨发育不全有关,其中主要为错义突变。HGMD已收录4种COL9A3基因突变导致假性软骨发育不全。鉴别诊断假性软骨发育不全、软骨发育不全、软骨发育低下三者均为短肢型矮小且临床表现相似,诊断有赖于骨骼x线表现及相关致病基因分析,假性软骨发育不全X线主要表现为SMED、软骨发育不全主要表现为SMD,三者鉴别详见下表:与多发骨骺发育不良MED鉴别:由COMP突变导致的显性MED,与假性软骨发育不全突变基因相同,两者最大的区别是MED没有脊柱受累。有时两者表型可有重叠。MED平均身高较PSACH高,退行性关节炎导致的疼痛出现较晚。与其他脊柱骨骺/干骺端发育不良鉴别:如SEDC、粘多糖病影像学有相应典型改变,改变不典型时,基因诊断可鉴别。治疗目前没有针对性药物治疗,不推荐生长激素单纯用于增高。手术治疗重点在于纠正双下肢畸形,发育期可行半骨骺阻滞术,其他可行截骨矫形术,但总体复发率高。由于PSACH患儿生长缓慢,香港大学深圳医院儿童骨科有开展半骨骺阻滞术联合生长激素治疗下肢畸形,可缩短总体治疗时间。对于下肢畸形严重的PSACH患儿,即使年龄不大,单纯半骨骺阻滞术也不适合,常需联合截骨内固定或截骨外固定架矫形。我们推荐畸形严重部位行截骨矫正,畸形不严重部位可行半骨骺阻滞术的杂交手术方式。因为骨骺受累,年龄大的患者常出现早发性骨关节炎,可予止痛对症治疗,成年后常需行关节置换术。颈椎融合固定用于颈椎失稳患者。腰椎前凸常由髋关节屈曲挛缩导致,一般行物理保守治疗。参考文献1.JohnA.Herring;Chapter40;Tachdjian‘sPediatricOrthopaedics,FifthEdition;2.JürgenW.Spranger,PaulaW.Brill,ChristineHall,GenNishimura,AndreaSuperti-Furga,SheilaUnger;Chapter1,2;Bonedysplasias:AnAtlasofGeneticDisordersofSkeletalDevelopment,FourthEdition;3.黎芳,麻宏伟,宋莹,etal.儿童短肢型遗传性骨代谢性疾病的临床分析及基因诊断.中国当代儿科杂志.2013;15(11):932-936.
 董忠信 主治医师 香港大学深圳医院 骨科88人已读
董忠信 主治医师 香港大学深圳医院 骨科88人已读
问诊记录 查看全部
- 患者:女 7岁8个月 软骨发育不良 最后交流时间 02.25软骨发育不良 董医生,您好,孩子的基因检测已经出来结果,麻烦您看看,感谢了 检查结果总交流次数49已给处置建议
- 患者:女 7岁8个月 双髋发育不良、软骨发育不良 最后交流时间 01.24双髋发育不良 软骨发育不良 医生,您好; 我简单描述下孩子情况,孩子在三岁的时候运动能力... 骨头边缘很毛躁,能否有治疗?总交流次数33已给处置建议
- 患者:男 2岁4个月 着急推门 宝宝站在门后脚指头被门夹到 流 最后交流时间 01.18着急推门 宝宝站在门后脚指头被门夹到 流 昨天晚上被夹的,流了很多血,大拇脚指甲盖中间有断裂的迹象,我... 一个多月前孩子被夹伤右脚大拇指,带去医院剪掉原伤甲片后做了消炎包扎处理,但新长出来的指甲中间仍然有一...总交流次数7已给处置建议
- schmid型干骺端软骨发育不全 目前孩子一岁8个月,一岁3个月会走路后发现O型腿严重,近期去... 药物研究进展及临床试验情况咨询总交流次数21已给处置建议
- 怀疑是骨骺发育无良 医生你好,我儿子我一直觉得身高比例不太对,手指脚趾都有点短,... 想问问孩子是不是要做什么检查?我很怕孩子是侏儒总交流次数13已给处置建议