




精选内容
-
蝴蝶宝贝 | 遗传性大疱性表皮松解症:症状、病因、诊断与治疗 作者 梅斯医学
大疱性表皮松解症(epidermolysisbullosa,EB)分为遗传性(先天性)和获得性(epidermolysisbullosaacquisita,EBA)两种。遗传性EB依据发病部位不同可分为三类:①单纯性大疱性表皮松解症(simplexEB,EBS),水疱在表皮内;②交界性大疱性表皮松解症(junctionalEB,JEB),水疱发生于透明层;③营养不良性大疱性表皮松解症(dystrophicEB,DEB),水疱发生在致密下层。遗传性大疱性表皮松解症是一组罕见的遗传性皮肤病,主要表现为皮肤或黏膜脆性增加,即受到轻微外伤或摩擦后出现破损、水疱或大疱改变。患儿皮肤像蝴蝶翅膀般脆弱,因此此病患儿又被称为蝴蝶宝贝。目前以皮肤伤口护理、保护及对症治疗为主,出现严重并发症者需要根据情况选择手术治疗。患者预后与疾病类型、基因突变位点及护理情况等有关。2018年5月11日,国家卫生健康委员会等5部门联合制定了《第一批罕见病目录》,遗传性大疱性表皮松解症被收录其中。1、一般概述大疱性表皮松解症(EB)是一种遗传性皮肤病,其临床特征是机械性创伤形成水疱。有四种主要类型,并确定了其他子类型。有一系列严重程度,在每种类型中,一种可能受到轻微或严重影响。EB的范围从需要修改某些活动的轻微不便到完全禁用,在某些情况下甚至是致命的。摩擦导致水泡形成。水泡可以在皮肤表面的任何地方形成,在口腔内,更严重的形式还可能涉及眼睛的外表面,以及呼吸道、胃肠道和泌尿生殖道。在某些形式的疾病中,会出现毁容性疤痕和致残的肌肉骨骼畸形。目前,没有治愈EB的方法。支持性护理包括日常伤口护理、包扎和疼痛管理。2、症状与体征大疱性表皮松解症分为四个亚型,每个亚型还都有亚型。1►单纯性大疱性表皮松解症(EBS)通常是显性遗传的,并且涉及角蛋白5和14以及plectin的基因紊乱。最近,也描述了几种基底上类型的EBS。出生时或出生不久有水疱、大疱,多出现在手、肘、膝、足等易摩擦部位。多数患儿在水疱愈合后还会有皮肤色素异常,肤色变浅或加深,但通常不会留下瘢痕或者皮肤萎缩。部分会出现口腔黏膜水疱或溃疡,但不太严重。甲营养不良、甲变厚、毛发稀疏等表现通常会缺如或程度较轻。EBS可能局限于手和脚,也可能是广泛分布,内部受累相对较轻。患有EBS的人可能会在手掌和脚底出现增厚的老茧,在婴儿期出现口腔水泡以及粗糙、增厚的手指甲/脚趾甲。EBS通常不会留下疤痕。有罕见的隐性形式。具有plectin突变的EBS可能与肌营养不良症有关。2►大疱性交界性表皮松解症(JEB)是隐性遗传的,涉及表皮和真皮之间连接的几个成分的基因突变,例如层粘连蛋白332(以前称为层粘连蛋白5)、plectin和a6b4整合素。有两种主要亚型,HerlitzJEB和JEB-other(包括非HerlitzJEB,以及伴有幽门闭锁的JEB和其他几种亚型)。连接HerlitzEB是由于三个层粘连蛋白332链中的任何一个发生突变,并且可能是非常严重的EB形式。由于严重感染(败血症)、营养不良、脱水、电解质失衡或阻塞性气道并发症,通常在婴儿期发生死亡。JEB-O的范围很广。口腔受累和牙齿表面的不规则点蚀在所有亚型中都很常见。患有幽门闭锁的婴儿在新生儿时期会出现喂养困难和腹胀,并在新生儿期表现为外科急症。3►营养不良性大疱性表皮松解症(DEB)营养不良型遗传性大疱性表皮松解症有常染色体显性遗传和常染色体隐性遗传两种遗传方式。并且涉及VII型胶原蛋白的缺陷。水泡出现在皮肤的下层,即真皮内。有两种主要的亚型,显性DEB(DDEB)和隐性DE(RDEB)。呈常染色体显性遗传的症状较轻,而呈常染色体隐性遗传的通常症状较重。显性营养不良EB(DDEB):DDEB通常是轻度的。起泡可能局限于手、脚、肘部和膝盖,也可能是全身性的。常见的发现包括疤痕、粟粒疹(微小的白色肿块)、黏膜受累以及指甲异常或缺失。一些家庭成员可能只有指甲营养不良。隐性营养不良EB(RDEB):RDEB通常比DDEB更普遍和更严重。除了疤痕、粟粒疹、黏膜受累和指甲营养不良外,常见的表现还包括营养不良、贫血、食道狭窄、生长迟缓、手指和脚趾的蹼或融合导致连指手套畸形(假性并指)伴功能丧失、挛缩、牙齿畸形、小口畸形和角膜擦伤。严重的广义RDEB(以前的Hallopeau-SiemensRDEB)往往是最严重的形式。4►Kindler综合征非常罕见,涉及皮肤的所有层,极度脆弱。出生不久后,皮肤受到摩擦后出现水疱和大疱;随年龄增长,水疱及大疱逐渐减少,皮肤部分情况变好,但同时皮肤开始出现明显的光敏反应,可能有光暴露部位皮肤异色改变。患者水疱愈合后可能有皮肤萎缩、指纹缺失、轻度并指等表现。3、病因遗传形式遵循常染色体显性或常染色体隐性遗传。还有一种罕见的获得性自身免疫性疾病,称为大疱性表皮松解症。编码表皮、基底膜或真皮中蛋白质的至少18个基因中的任何一个发生突变都会导致皮肤完整性差,从而导致脆弱。隐性遗传疾病发生在个体遗传了同一性状的异常基因的两个拷贝时,每个拷贝来自父母一方。如果一个人接受一个正常基因和一个疾病基因,该人将成为该疾病的携带者,但通常不会出现症状。每次怀孕,两个携带者父母都通过缺陷基因并有受影响的孩子的风险是25%。每次怀孕生下一个像父母一样是携带者的孩子的风险是50%。一个孩子从父母双方那里获得正常基因并在该特定特征上遗传正常的机会是25%。男性和女性的风险相同。所有个体都携带至少4-5个异常基因。近亲(近亲)的父母比无关父母携带相同异常基因的可能性更高,这增加了患隐性遗传疾病的孩子的风险。当只需要一个异常基因的一个拷贝来引起特定疾病时,就会发生显性遗传疾病。异常基因可以遗传自父母中的任何一方,也可以是受影响个体中新突变(基因改变)的结果。每次怀孕将异常基因从受影响的父母传给后代的风险为50%。男性和女性的风险相同。EBS与编码角蛋白5和14的基因突变有关;JEB与编码板层素5、ⅩⅦ型胶原(BPAG2)等物质的基因突变有关;DEB与编码Ⅶ型胶原的基因(COL7A1)突变有关。由于编码表皮和基底膜带结构蛋白成分的基因突变,使这些蛋白合成障碍或结构异常,导致不同解剖部位水疱的产生。目前本病已知有20个不同致病基因,其中第19个致病基因KLHL24是我国学者林志淼及杨勇等发现。4、流行病学估计每50,000名活产婴儿中就有1名出现某种类型的EB。这种疾病发生在全世界的每个种族和族裔群体中,并且平等地影响两性。目前预估中国约有1200名被称为"蝴蝶宝贝"的EB患者。5、鉴别诊断新生儿期的任何水疱疾病都可能与EB相似。这些包括单纯疱疹病毒、表皮松解性鱼鳞病、大疱性脓疱病和色素失禁。6、诊断应避免根据新生儿期的表现对EB类型进行临床诊断,因为该年龄组中所有类型的EB可能看起来相似。当怀疑EB时,应进行皮肤活检并将其送到适当的实验室,以通过透射电子显微镜(TEM)和/或免疫荧光抗体/抗原作图确认诊断。大多数已知与各种类型EB相关的基因突变的分子遗传学检测在临床上是可用的。7、治疗根据定义,遗传性EB是一种以皮肤显着脆弱为特征的遗传传播疾病。任何创伤,无论看起来多么轻微,都可能导致EB儿童或成人的皮肤撕裂或起水泡。以下是避免或最小化此问题的推荐方法:1.减少摩擦:在处理任何EB患者的皮肤时都应格外小心。2.非粘性绷带和敷料:不应在皮肤表面使用粘性或半粘性敷料、绷带、创可贴或胶带。相反,应该用适当的非粘性敷料覆盖伤口,然后用卷起的纱布进一步松散包裹。这可以通过使用管状敷料保持器来固定。3.保持皮肤凉爽:EB患者的皮肤上不应涂抹任何热的东西。特别是洗澡水的温度不应高于体温。患者应避免长时间暴露于环境高温和潮湿环境中。如果可能,应尽可能寻找有空调的环境。4.管理水泡:因为EB中的水泡不是自限性的,并且可以充满液体并长得很大,所以需要将它们排出。5.服装:对于年幼的孩子,尿布可能需要在腿部和腰部添加额外的衬垫。只要有可能,应穿宽松的衣服。如果衣服的接缝处出现水泡,衣服可能会从里到外穿着,标签、袖口和领口可能会被去除。宽松的软垫鞋通常具有更好的耐受性。6.营养缺乏:许多患有EB的儿童由于伤口慢性失血、营养摄入不足、铁吸收不良和慢性炎症导致的骨髓抑制而变得贫血。与在照顾特殊需要患者方面经验丰富的营养师合作非常重要。通常需要治疗缺铁性贫血。其他患者有硒和肉碱或维生素D缺乏症,这可能使他们易患心肌病和骨质疏松症。许多患者无法茁壮成长,需要进行胃造口术。7.癌症监测:鳞状细胞癌是EB死亡的主要原因,通常发生在生命的第二个十年之后。RDEB和JEB患者在其一生中患皮肤癌的风险增加。所有EB患者至少每年对所有皮肤区域进行检查是非常重要的。8、预后预后差异较大,取决于遗传性大疱性表皮松解症亚型和患者的健康状况,轻型遗传性大疱性表皮松解症随年龄增加症状会得到改善,重型患者很可能出现各种严重并发症,多数严重威胁患者功能及影响寿命。极重型患者常在婴幼儿期死亡。
周飞红医生的科普号 2022年08月26日
2022年08月26日 275
275
 0
0
 1
1
-
大疱性表皮松解症
大疱性表皮松解症(Epidermolysisbullosa,EB)是一组罕见的遗传性疾病,分为遗传性和获得性两种,以皮肤和黏膜对机械损伤易感,而形成大疱为特征的慢性非炎症性大疱性疾病。其在临床上和遗传学方面具有异质性,特征是上皮组织明显的机械脆性,轻微创伤后即出现水疱、血疱和糜烂。遗传性大疱性表皮松解症好发于婴儿期,获得性大疱性表皮松解症好发于老年人。大疱性表皮松解症(EB)最可靠的患病率和发病率数据来自美国国家大疱性表皮松解登记研究,在16年的时间里(1986-2002年),EB的患病率估计为11/100万人,发病率约为20/100万活产。在同一时期,EB的亚型中,单纯性EB的发病率约为8/100万活产,交界性EB的发病率约为3/100万活产,显性遗传营养不良性EB的发病率约为2/100万活产,以及隐性遗传营养不良性EB的发病率约为3/100万活产。一、疾病分类大疱性表皮松解症分为遗传性和获得性两类。1、遗传性大疱性表皮松解症:又分单纯型、交界型、营养不良型三种。⑴单纯型大疱性表皮松解症:多为常染色体显性遗传,最轻型,愈后一般不留瘢痕。通常在1岁内发病,水疱在表皮内。⑵交界型大疱性表皮松解症:罕见,为常染色体隐性遗传,水疱发生于透明层。⑶营养不良型大疱性表皮松解症:水疱发生在致密下层,分为常染色体显性和隐性遗传两种。显性遗传营养不良型大疱性表皮松解症,在婴儿早期或儿童期发病,损害愈合缓慢,治愈后留有萎缩性或增殖性瘢痕,隐性遗传营养不良型大疱性表皮松解症患者病情更重。2、获得性大疱性表皮松解症:是一种少见的自身免疫性疾病,临床上多见于成年人,儿童和老年人也可发病。考虑与许多系统性疾病有关,如炎症性肠炎、类风湿关节炎、糖尿病、淀粉样变、系统性红斑狼疮和血友病等。二、发病原因遗传性大疱性表皮松解症(EB)由于编码表皮和基底膜带结构蛋白成分的基因突变,使这些蛋白合成障碍或结构异常,导致不同解剖部位水疱的产生。主要病因为:单纯性大疱性表皮松解症,与编码角蛋白5和14的基因突变有关。交界性大疱性表皮松解症,与编码板层素5、ⅩⅦ型胶原等物质的基因突变有关。营养不良性大疱性表皮松解症,与编码Ⅶ型胶原的基因突变有关。获得性大疱性表皮松解症是一种自身免疫性疾病。病因不明,有研究发现与体内产生抗Ⅶ型胶原抗体和HLA-DR2阳性有关。诱发因素:皮肤受到摩擦和碰撞可诱发大疱性表皮松解症。三、临床症状各型大疱性表皮松解症的共同特点为皮肤脆性增加,即或黏膜轻度摩擦后即出现水疱、大疱。皮损好发于手、足、肘、膝、臀部等摩擦部位,也可泛发全身,有的伴有瘢痕、萎缩、甲营养不良或皮肤外表现,重者可致残或致死。各型大疱性表皮松解症的典型症状:1、遗传性大疱性表皮松解症⑴单纯型大疱性表皮松解症:常在受压或机械损伤后发生,在大疱出现前,受累皮肤可出现淡红斑、轻度瘙痒或烧灼感,以后发生清澈紧张的大疱,偶见血疱。大疱破裂形成糜烂,但易愈合。无感染并发的情况下,不留疤痕。手、足、膝、肘、颈等为好发部位,尤以手足最多。约2%的患者口腔、生殖器、肛周黏膜可轻度受累,一般身体发育情况正常。⑵交界型大疱表皮松解症:出生后即有广泛的水疱、大疱、糜烂和结痂,愈后出现萎缩性瘢痕,口腔黏膜常有糜烂、溃疡、瘢痕使张口困难。患者一般情况差,生长发育迟缓,常有严重贫血,预后差,大多数患者在2岁内死亡。⑶营养不良型大疱表皮松解症:分为常染色体显性和隐性遗传两种。显性遗传营养不良型大疱表皮松解症好发于四肢,表皮囊肿和粟丘疹常见,少数病例累及黏膜。隐性遗传营养不良型大疱表皮松解症患者病情更重,出生时出现泛发性水疱、大疱和糜烂,有的为血疱,尼氏征(+),皮损缓慢愈合后留下萎缩性疤痕及粟丘疹,严重的瘢痕常造成膝、肘、腕、踝关节屈曲挛缩,功能障碍。黏膜受累,以食管复发性大疱最为常见、严重。食管病变发生在幼儿期,但在成人期表现明显,吞咽困难,常导致吸入性肺炎。往往因泛发糜烂造成患者体液及蛋白质丢失,多在儿童期或青春期因继发感染、败血症、肺炎或营养不良而死亡。2、获得性大疱性表皮松解症多发生在老年人,皮损好发生在手指、足、肘、膝关节侧面,容易受外伤的部位,皮损为无炎症反应的皮肤上形成水疱、大疱、糜烂等损害,愈后可留萎缩性瘢痕,可见粟丘疹,部分病人伴有毛发、甲损害,以及黏膜损害。并发症营养不良型大疱表皮松解症50%~80%患者在慢性糜烂区域发生鳞癌,容易侵袭和转移。交界型大疱性表皮松解症,常合并有气管水疱、狭窄或阻塞。生长发育迟缓,常有严重贫血。患儿常死于败血症、多器官衰竭和营养不良。四、临床检查1、组织病理检查:镜下可见基底细胞水肿、液化变性,表皮下疱,疱内及真皮浅层偶有炎症细胞浸润,透射电镜可进一步确定水疱的精确位置。2、血液检查:若有贫血时,外周血象可见红细胞计数和血红蛋白量下降。并发感染时,外周血象白细胞计数和中性粒细胞计数显著升高;重症感染伴有水电解质紊乱时,应做血电解质、pH值和肝肾功能、免疫荧光抗原定位等检查。有感染时,同时做分泌物培养及药敏。3、基因检测:此病具有遗传性,因此可通过基因检测进行诊断。4、免疫荧光检查:可初步确定可疑致病基因。5、其他检查:重症病例应做胸部X线片、心电图、B超等检查。五、诊断大疱性表皮松解症根据典型病史、家族史、临床特点,结合免疫组化及透射电镜检查一般可以确诊及分型。⑴单纯型:系显性遗传,皮损为大小不等的大疱和水疱,水疱在表皮内,无棘层松解征,愈后不留瘢痕,可留有暂时性色素沉着。受累患儿生长发育正常,毛发、甲、齿、黏膜很少受累,至青春期可获改善。⑵交界型:常染色体隐性遗传,出生后就有广泛水疱、大疱、糜烂,愈后有萎缩性瘢痕,口腔黏膜可受累,水疱发生于透明层。⑶营养不良型:损害为松弛的大疱,水疱发生在致密下层,棘层松解征阳性,愈后留下萎缩性瘢痕,常伴发粟丘疹,有色素障碍。身体和智力发育正常,毛发、牙齿常不累及,有时有鱼鳞病、毛周角化症、多汗或厚甲等。⑷获得性:可由药物、感染、卟啉病,淀粉样变等引起,常伴有相关疾病的其他表现。六、鉴别诊断1、新生儿脓疱疮:极易传染,可呈流行性。水疱易破裂,内容迅速变为脓性,可查见葡萄球菌或链球菌,炎症显著,易于治愈。2、皮肤卟啉病:水疱多见于背、面部、耳等曝光部位,对光敏感。可见多毛,常伴发肝损害。尿及粪中尿卟啉及粪卟啉增高。3、儿童线状IgA大疱性皮病:发病不限于摩擦部位,无遗传史,愈后不留萎缩性瘢痕。直接免疫荧光检查可见IgA沿基底膜带呈线状沉积。4、新生儿天疱疮:往往泛发全身,疱壁松弛,组织病理为棘层松解所致的表皮内水疱,免疫病理显示角质形成细胞间IgG、IgA、IgM或C3网状沉积,血清中存在针对桥粒成分的天疱疮抗体。5、大疱性丘疹性荨麻疹:常伴有明显的瘙痒,且有水肿性丘疹存在。七、临床治疗大疱性表皮松解症目前尚无特效疗法,因为在出生时或婴儿早期发病,大多由创伤引起,患儿应保护皮肤,避免外伤、摩擦,防止继发感染。可用非粘连性合成敷料、无菌纱布或广谱抗生素软膏防治感染。生活在凉快的环境中,并避免高温对一些患者是有益的。㈠西医治疗1、药物治疗可选用维生素E100mg,3次/d和枸橼酸钠2g,3次/d。严重患者特别致死型患者,可应用皮质类固醇激素,新生儿开始剂量为140mg/d,分数次服用。合理外用抗生素,如莫匹罗星或夫西地酸,并应在每2~6周轮换不同的药物,以尽量减少诱发细菌耐药。抗生素可用于预防或控制继发感染。严重患者特别是致死型,贫血严重者,常需输血或其他支持疗法。有明确感染的伤口通常需要全身性抗生素治疗,应根据细菌培养和药敏试验的结果来选择抗生素治疗。对于轻至中度疼痛,可以单独应用镇痛剂(如扑热息痛、对乙酰氨基酚),也可以与非甾体抗炎药合并使用。对于重度疼痛的治疗,可能需要应用阿片类药物或者抗焦虑药物。苯妥英钠是治疗营养不良型大疱性表皮松解症有希望的药物,该药能抑制皮肤的胶原酶,初用5mg/(kg·d),分数次服,逐渐加量到8mg/(kg·d),半年后减为6mg/(kg·d),1年后改为1天量隔天服。2、手术治疗⑴分指手术:大疱性表皮松解症患者的手畸形类似于临床上常见的烧伤后瘢痕挛缩畸形,可采取分指手术彻底分离、松解粘连及屈曲的手指。⑵食管黏膜扩张术:适用于食管挛缩、狭窄者。3、营养治疗所有重型大疱性表皮松解症(EB)患者均有营养受损并且需要营养支持,贫血患者需要补充铁,同时给予或者不给予促红细胞生成素。有证据显示,骨质减少或骨质疏松的患者应补充钙和维生素D。4、其他治疗大疱性表皮松解症伤口敷以不粘性硅敷料,能吸收渗出液的泡沫敷料和不粘性硅胶带。在严重细菌定植的伤口中,稀释的漂白剂洗浴或湿敷法,局部用抗菌剂和局部用抗生素可用于减少细菌载量。5、基因治疗单纯型大疱性表皮松解症多为常染色体显性遗传,使基因治疗更加困难,因为突变体等位基因的表达必须是隐性的。目前的方法是通过同源重组技术去除患者的缺陷等位基因。尽管用患者的活检皮肤做基底角质形成细胞培养是可行的,但同源重组频率极低。准确地了解致病突变基因和在mRNA、蛋白水平这些突变引起的功能改变是开发用基因治疗营养不良型大疱性表皮松解症的必要前提。可以相信数型大疱性表皮松解症是基因治疗的可行性对象,包括RDEB。然而,由于Ⅶ型胶原的mRNA分子大(大于9.2kb),常规的通过病毒转导把野生型cDNA导入患者的角质形成细胞很不易成功。因此,已经寻求其他办法,包括通过生物溶解颗粒导弹方法(biolisticparticlebombardment)直接把遗传物质导入皮肤细胞;通过口标性转拼使用核酶介导的突变mDNA修复和应用嵌合的RNA/DNA寡核苷酸进行靶基因校正。这样,阐明不同型大疱性表皮松解症的基因基础为将来用基因治疗方法治疗这些致残性皮肤病奠定了必要的基础。数种较轻亚型的交界型大疱性表皮松解症也是基因治疗的可行性对象,包括非致死型交界型大疱性表皮松解症如GABEB。将来,向患者角质形成细胞内用体外和体内方法导入相对小的野生型cDNA(BPAG2和ITCB)很可能要比大一点的mRNA易成功。㈡中医疗法1、脾虚湿盛型治法:健脾除湿。方药:白术10g,茯苓10g,薏米15g,陈皮10g,枳壳10g,车前子10g,泽泻10g,茵陈10g,竹叶6g,灯心2g,冬瓜皮lOg。水煎服,日一剂。也可服人参健脾丸。加减:气虚明显者加党参,继发感染者加蒲公英、银花。2、脾肾阳虚型治法:温补脾肾。方药:黄芪10g,党参lOg,白术10g,茯苓10g,熟地10g,巴戟天10g,菟丝子10g,桂枝10g,肉桂l~2g,当归lOg,阿胶lOg。也可服用补中益气丸加四神丸或服全鹿丸。3、局部治疗⑴地榆油:取生地榆适量油炸去渣外涂患处,或制成消毒油纱布,每天换药1次。⑵清凉膏(赵炳南方):当归30g,紫草6g,大黄面5g,香油300g,黄蜡120g。当归、紫草浸入香油3天后,用微火熬至焦黄,离火滤清,入黄蜡加火熔匀,等冷后加大黄面搅匀成膏,外敷患处。本方具有清热解毒,凉血止痛功效。⑶祛湿散、甘草油(赵炳南方)调为糊状外敷。祛湿散由川黄连、川黄柏各24g,黄芩14g,槟榔96g组成,共为细末备用,甘草油为甘草30g,香油300g,油炸去渣即成。八、护理大疱性表皮松解症的患者日常生活中应注意保护皮肤,避免皮肤受到摩擦、压迫等刺激,并保持皮肤清洁,避免出现继发感染。要有规律的作息,避免过度劳累,不能熬夜。九、饮食调理大疱性表皮松解症在饮食上无特殊注意,建议有规律的高蛋白饮食,多进食鸡蛋、鸡肉、奶制品等营养机体、提高免疫力。注意补充维生素类食物,如新鲜水果、蔬菜、谷类、动物的肝脏等。饮食要清淡规律,避免辛辣、油腻、刺激性及引起过敏反应的食物,例如辣椒、烟酒、海产品,忌浓茶、咖啡,避免高脂、高糖饮食。十、预后就特定死因和风险等级来讲,不同亚型患者的死亡风险差异很大。如果给予细致积极的治疗与护理,出生后能存活12~24个月的大多数患儿至少可以活到成年。尤其是伤口护理方面的进步,已大大降低了具有广泛伤口感染患者死于脓毒症的风险。⑴单纯型大疱性表皮松解症(EBS),尽管一些患者水疱非常严重,但很少危及生命。因局限性皮肤屏障功能丧失,易于继发感染。疱疹样大疱性表皮松解症是最严重的一型。因为水疱裂隙位于表皮内,愈后不留瘢痕。患者指(趾)甲可能脱失,但通常可再生。⑵交界型大疱性表皮松解症(JEB)的Herlitz型,又称为致死型和gravis型,常不能存活过婴儿期,40%多在出生后l年内夭折。是所有大疱性表皮松解症中最严重的一型。⑶营养不良型大疱性表皮松解症的隐性遗传型临床表现多样。严重型损害具致残性,称为Hallopeau-Siemens(TTS-RDEB)型。ITS-RDEB最严重的合并症是在慢性糜烂区域发展为鳞状细胞癌。高于50%的TTS-RDEB患者在30岁左右时发展为此癌,许多死于癌转移。Bart综合征为DDEB的临床亚型预后较好。
李希新医生的科普号 2022年01月22日709
0
3
2022年01月22日709
0
3
-
痒疹型大疱性表皮松解症的新治疗
什么是痒疹型大疱性表皮松解症?为什么会引起痒疹型大疱性表皮松解症?痒疹型大疱性表皮松解症(epidermolysis bullosa pruriginosa,简称EBP)是一种罕见的遗传病,是营养不良
林志淼医生的科普号 2021年03月03日6718
3
11
2021年03月03日6718
3
11
-
遗传性大疱性表皮松解症,患者后期除了特别容易出现皮肤癌,还很容易因为手指、脚趾的粘连而致畸、致残。
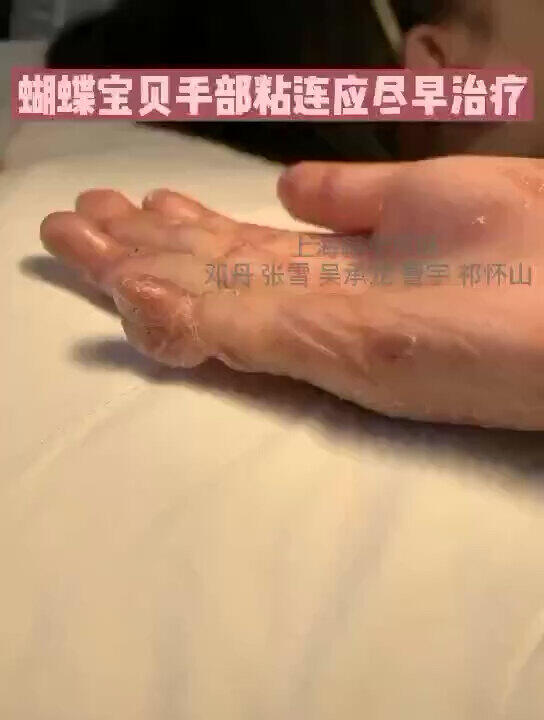
 邓丹医生的科普号
邓丹医生的科普号 2020年08月25日1583
0
1
2020年08月25日1583
0
1
-
2019年大疱性表皮松解症新分类方法 译文
通讯作者:Cristina Has(cristina.has@uniklinik-freiburg.de)网址为doi:10.1111/BJD.18921关于这个话题我们知道些什么?·大疱性表皮松解症是一组伴有皮肤起泡的遗传性疾病。·关于诊断和分类的最后一次更新建议已于2014年发布。这项研究增加了什么?·我们引入了皮肤脆性遗传性疾病的概念,经典的EB代表了这一概念的原型。本文就EB的临床和遗传学方面、基因型-表型相关性、疾病修饰因素和自然病史等方面进行综述。·其他皮肤脆性疾病,如脱皮疾病、糜烂性疾病、过度角化疾病和结缔组织疾病皮肤脆性,被单独归类。从医疗和社会经济提供护理的角度考虑,这些“EB相关”疾病应该在EB的保护伞之下。摘要背景:自2014年关于大疱性表皮松解症(EB)的最后一次国际共识会议的报告发表以来,已经发现了几个新的基因和临床亚型。目的:基于新的临床和分子数据,我们试图对皮肤脆性疾病进行重新分类,重点放在EB上。方法:这是一项协商一致的专家评审。结果:在这份最新的共识报告中,我们引入了皮肤脆性遗传性疾病的概念,经典EB代表了这一概念的原型。其他皮肤脆性的疾病,其中水泡是临床表现的一小部分,或者因为皮肤裂隙非常浅而看不到,被归类为单独的类别。这些疾病包括脱皮疾病、浸渍性疾病、过度角化疾病和伴有皮肤脆性的结缔组织疾病。由于皮肤脆性的常见表现,这些“EB相关”疾病应该在EB保护伞下考虑医疗和社会经济方面的护理。结论:提出的分类方案对临床医生和研究人员都有一定的参考价值,既强调EB的临床特征,也强调EB的遗传学特征。引言遗传性皮肤脆性疾病(SF)的特征是结构异常,降低了皮肤对机械应力的弹性。根据皮肤内分子和结构缺陷的位置,临床表现可能包括脱皮、水泡、糜烂、溃疡、伤口或疤痕。2019年4月,一些领先的专家在伦敦会面,审查相关数据,修订这些疾病的分类系统,特别考虑大疱性表皮松解症(EB),并尽可能侧重于分子病因学。EB是一组典型的SF疾病,定义为皮肤-表皮交界处断裂的微小机械损伤形成水泡(图S1,表1)1四种主要的经典EB类型是:单纯EB(EBS)、交界性EB(JEB)、营养不良性EB(DEB)和Kindler EB(KEB)。其他与SF有关的疾病,其中水泡只是临床图像的一小部分,或者因为皮肤劈裂非常浅而看不到,被归类为单独的类别。这些疾病包括皮肤脱皮疾病、糜烂性疾病、角化过度疾病和SF结缔组织疾病(表2)。由于SF的常见表现,在鉴别诊断时应考虑到这些“EB相关性”疾病。建议的系统仍然主要面向临床,因为SF患者的分类是从床边开始的,基于个人和家族史,以及有没有特定的临床特征。只有在以后,实验室诊断才能根据分子结果对这些患者进行更准确的细分(表3-5)。EB分类很复杂,因为同一基因中的突变可能以常染色体显性或隐性方式遗传,并可能导致不同的临床表型(如Krt5、KRT14、PLEC、COL17A1或COL7A1)。另一方面,在DEB和EBS中,相似的表型可能是显性的或隐性的,也可能是由不同基因(如COL7A1、Krt5、KRT14、PLEC、DST、EXPH5或KLHL24)的突变引起的。如果怀疑EB,应该在早期进行免疫荧光测绘和分子遗传学诊断,以确定准确的亚型,改善预后,并使遗传咨询、产前诊断、纳入临床试验和精确医学成为可能2,3。最近出版了EB实验室诊断指南2,因此不会在此文中讨论。结合临床和分子方面,以前介绍的“洋葱皮”亚型分类方法1,包括主要的EB类型(基于皮肤分裂水平)、遗传方式以及临床和分子特征被证明是有用的,并被进一步推荐。综合征性SF障碍的概念是最近提出的4,包括以胃肠道或泌尿生殖道、心肌、骨骼肌等其他器官或系统为主要表现的疾病。(表S1)。与此形成对比的是,严重EB亚型伴随着大面积持久的皮肤和粘膜缺损,特别是严重的隐性DEB(RDEB),会演变为继发性皮外并发症5,6。经典EB的类型典型EB的主要临床和遗传特征在辅助资料附录1中描述。临床方面在图1-5和附录2(图S2-S4)中说明。单纯性大疱性表皮松解症是由角质形成细胞基底层内的裂隙引起的皮肤起疱定义的单纯性大疱性表皮松解症。在大多数情况下,EBS是以常染色体显性方式遗传的;常染色体隐性遗传在西方国家很少见,但在世界一些地区相当常见7,8。临床严重程度广泛,从脚上轻微起疱,到皮外受累和致命的亚型。遗传背景是复杂的,有七个不同基因的突变。新的基因,KLHL249,10和CD15111,自上一次分类以来已经被发现,并扩大了EBS的范围;仍然有一定比例的病例在遗传上仍未解决。EBS是最常见的EB类型,大多数轻度病例仍被低估。来自美国的数据显示,总发病率为每百万活产7.87例,流行率为每百万例6例。常见的EBS亚型是由编码角蛋白5或14的基因内的单等位基因突变引起的,包括:局限型(以前称为Weber-Cockayne)、中度型(以前称为中度泛发型或Kbner)和重度(以前称为重度泛发型或Dowling-Meara)EBS。EBS亚型中的罕见类型的具有临床异质性,包括几个综合征疾病(表3)。从遗传学上讲,这些疾病要么是常染色体显性的,要么是隐性的,其中一些是由特定的突变引起的,这些突变具有不同的分子和表型后果,目前机制还不完全清楚。已经报道了几个由ITGB4或COL17A1(通常与JEB相关的基因)突变引起的EBS病例,这些突变破坏了各自蛋白质的细胞质结构域13,14。交界性大疱性表皮松解症是一种常染色体隐性遗传病,其特征是皮肤起水泡,并在皮肤基底膜区(BMZ)的透明层形成裂隙。严重程度在两个主要亚型中差别很大,中度和重度,后者与生命最初6-24个月的早期死亡有关。流行病学数据表明,与单纯型或营养不良型EB相比,JEB较少见。来自美国的数据显示,总发病率略高于每百万活产2人,然而,低于这一数字的患病率可能反映了重症的寿命较短。JEB的两个主要亚型是重度JEB(以前称为泛发型JEB,Herlitz JEB)和中度JEB(以前称为JEB中度泛发型,非Herlitz JEB)。虽然编码层粘连蛋白332亚单位链的三个基因(LAMA3、LAMB3、LAMC2)中的一个基因的双突变会导致这两种形式中的任何一种,但XVII型胶原基因(COL17A1)的双等位基因突变也可以导致中度和罕见的重度JEB表型。营养不良性大疱性表皮松解症DEB的特征是真皮最浅处致密层下方出现皮肤裂隙。在超微结构上,这与锚定纤维的水平相对应,反映了编码这些结构的主要成分VII型胶原基因潜在的分子病理。DEB可显性或隐性遗传,一般情况下,RDEB比显性病(DDEB)严重,但类型间有相当大的表型重叠。DEB的特点是在皮肤和各种粘膜起泡后形成疤痕。在DEB愈合的水疱处,粟丘疹是特定的特征。继发性皮外并发症在较严重的RDEB中很常见。对DEB发病率和流行率的估计各不相同,反映了不同国家在招募患者队列方面的差异。据报告,挪威和美国的DDEB发病率分别为1.4‰和2.5‰17,美国的RDEB发病率为3.05‰。美国15和西班牙18所有类型DEB的流行率估计约为6‰,澳大利亚为188‰,苏格兰为20‰20,后者可能反映了更多的捕获,而不是真正的更高流行率。DEB的所有亚型,无论是显性的还是隐性的,都是由编码VII型胶原COL7A1的基因突变引起的,COL7A1是皮肤BMZ锚定纤维的主要成分。DEB的主要亚型包括局限型DDEB(以前仅包括指甲、胫骨前和肢端DDEB)、中度DDEB(以前称为泛发型DDEB)、中度RDEB(以前称为RDEB中度泛发型,非Hallopeau-Siemens RDEB)和重度RDEB(以前称为RDEB重度泛发型,Hallopeau-Siemens RDEB)。发现了一些较罕见的DEB形式(表5)。Kindler EBKindler EB(KEB)是一种罕见的EB类型,自1954.21首次描述以来,全世界报告了约250名受影响的个体,它在孤立或血缘关系的人群中更为常见22,23。为避免对这种疾病的症状性质的混淆,建议将Kindler EB命名为Kindler EB,而不是Kindler综合征。遗传基础表现为FERMT1(syn.。KIND1),编码fermitin family homologue 1 1(kindlin-1),这是一种局部粘连的细胞内蛋白。皮肤脆性的其他疾病除了经典的EB亚型外,SF是其他遗传性疾病的特征,包括脱皮、糜烂、过度角化和结缔组织疾病(表2)。这些疾病在皮肤和/或皮肤屏障缺陷存在和致病机制方面与EB相似24,在鉴别诊断中应加以考虑,特别是在新生儿中。因此,我们建议在EB的下一代测序panel中包括相应的基因。表S3-S5总结了包括在这些组中的疾病的主要临床和分子特征。有关详细说明,请参阅原始文章和评论文章25-31。与SF有关的几种疾病需要更详细的说明。据报道,肢端脱皮皮肤病在婴儿中类似于局限性的EBS,而在成人中,肢体上的特征性脱皮可以进行临床诊断32,33。由于桥粒缺陷引起的棘皮松解的糜烂性疾病,可能表现为浅层水泡,但大多数是糜烂。角化性鱼鳞病的个体在出生和婴儿时表现出皮肤起泡,但角化过度症很快变为主要临床表现。到目前为止,已在单个个体中描述了一种口腔粘膜棘突松解性水泡的疾病,其原因是桥粒蛋白3基因(desmoglein 3 gene.)的纯合子无义突变34。然没有被列为“经典”EB,但这组疾病在皮肤上很明显,通常粘膜脆性是关键的表型特征,带来了同样的临床负担和医疗需求。因此,就医疗和社会经济提供护理而言,他们应该被视为EB保护伞下的成员。EB的基因型-表型相关性研究与经典EB和其他皮肤脆性疾病相关的致病变异数量正在稳步增长,达到数千种(人类基因突变数据库(HGMD),专业)。虽然存在许多个体变异和基因型-表型关系,但仍有一些一般规则适用,如下所述。它们的重要性取决于它们的医学相关性,在预测新生儿疾病严重程度的背景下,以及确定基因测试的优先顺序以节省资源。在对患者及其家属进行咨询时,重要的是要意识到这些相关性的局限性,因为已经报道了这些规则的许多例外情况。EBS与Krt5和KRT14突变的基因型-表型相关性对于具有Krt5或Krt14致病变异的常染色体显性EBS,受影响的氨基酸在角蛋白多肽中的位置决定了表型的严重程度,并可以进行预测。螺旋起始区或终止模体中高度保守的氨基酸的替换会削弱角蛋白5和14多肽的异二聚化,并导致严重的EBS,而基因其他区域的替换会导致局灶性EBS(www.interfil.org)。单等位基因的框内缺失、剪接位点或终止密码子(PTC)突变通常会导致截短蛋白的形成,并具有显性的负性影响。角蛋白5或14的一些致病变异与非常严重的临床病程相关38、39,常染色体隐性遗传性EBS大多是由KRT14无义或移码致病变异引起的。据报道,有两例角蛋白5缺失,均为致死性40,41。JEB基因型-表型相关性研究导致层粘连蛋白332(laminin 332)或整合素α6β4(integrinα6β4)缺失的致病变异与早期致死性有关42,43,而大多数COL17A1致病变异导致XVII胶原缺失,但与不那么严重的表型相关44。允许表达残留蛋白量的错义或剪接突变导致较温和的表型。对JEB患者的观察清楚地表明,即使截短并推测部分功能,仅有5%-10%的残留蛋白也能显著减轻表型(44,45篇综述)。特别令人感兴趣的是一些与自我改良的JEB相关的致病变异,其表型比预期的要温和。潜在的分子机制是剪接的交替调节、PTCs的自发通读或含有PTCs的外显子的跳过46-48。DEB的基因型-表型相关性DDEB主要是由于COL7A1外显子73的VII型胶原铰链区周围胶原区的甘氨酸取代所致49,最常见的是p.G2043R。然而,即使在同一个家庭中,携带相同甘氨酸替代物的个体之间也有相当大的临床变异性。此外,VII型胶原三螺旋中的一些甘氨酸替代突变与RDEB相关,另一些突变可能导致显性或隐性DEB51.单等位基因剪接位点或插入突变导致整个外显子(如外显子87)52、或甚至三螺旋结构域53内的大片段缺失导致轻度局灶性DDEB。RDEB是由广泛的致病变异引起的,导致缺乏VII型胶原。COL7A1显性和隐性突变的复合杂合性已多次被报道与严重的DEB有关54。自我改善的DEB与外显子(例如外显子36)55的框内跳跃或与特定的甘氨酸替代有关56,57。COL7A1中特定的甘氨酸和精氨酸替代突变与反向RDEB有关,提示它们可能影响VII型胶原的热稳定性58.疾病修饰因子在某些情况下,偏离预期的基因型-表型相关性可以通过遗传或表观遗传修饰因子的参与来解释。一种类型的遗传修饰物由顺式基因中的变异体表示,其可能让相应等位基因的表达,导致包含致病变异的外显子的框内跳过59。这样的事件可以减轻疾病的严重性,因为截短的分子通常会保留部分功能。第二种类型的遗传修饰物是嵌合体,或者作为致病变体的合子后嵌合体(描述为COL7A1和PKP1)60-62,或者作为回复嵌合体(revertant mosaicism)(描述为KRT1463,64,COL17A165-67,LAMB368,COL7A169-71,和FERMT172,73)。显性突变的合子后嵌合可能解释了父母的表型明显较轻,而后代的疾病更严重,而受影响皮肤的Blaschko线形区域可能是第二次隐性突变的嵌合造成的。据报道,在所有类型的EB中都有反复性嵌合体,并解释了由于致病变异的自发修复而改善了机械稳定性的皮肤区域。第三,两个EB相关基因,如Krt5和KRT1475,EXPH5和COL17A176或PLEC1和ITGB477的双基因突变已被报道导致意外的表型。第四种类型的遗传修饰机制由与EB没有直接关联的基因中的变异所代表,但它们的产物可能调节或影响EB相关的蛋白。这样一个例子是MMP1,它编码基质金属蛋白酶1,这是一种降解VII型胶原的酶。据报道,MMP1启动子中的一个频繁的功能性遗传变异与RDEB中更高的疾病严重程度有关,这是由于VII型胶原合成和降解之间的不平衡78。最后,在血缘关系的背景下,EB和其他遗传疾病的共存导致了复杂的、显然是“新的”表型。调控基因表达的表观遗传因子包括异染色质成分、多梳蛋白、非编码RNA和DNA甲基化80;这些机制在EB中仍有待证实.81,82。RDEB报道了核心蛋白聚糖和转化生长因子-b基因表达的改变,它们要么是慢性创面愈合过程中产生的继发性效应,进一步恶化了局部皮肤环境,要么是由离散的遗传变异引起的。然而,它们代表着潜在的治疗目标。其他表观遗传但未知的因素仍有待确认。最后,个人(如个性、家庭背景)、社会经济(如获得医疗保健和卫生条件)和环境因素(如气候)对EB的进程有重要的影响。综上所述,遗传、表观遗传和非遗传修饰因素似乎对EB表型有很强的影响;这种变异性意味着表型通常反映一个连续体,因此,严格划分亚型并不总是简单的。自然病程不同类型EB的临床特征和并发症往往会随着时间的推移而变化和演变,了解这一点对于识别不同的亚型并预测临床过程和相关问题是非常必要的。虽然这一自然历史在一定程度上反映了生命中不同发育阶段的变化,但某些EB亚型具有自然进化,其严重程度不同,要么恶化或改善,要么随着时间的推移发展或丧失特定特征。根据临床特征区分新生儿期EB的主要类型是极其不可靠的,并突出了快速和准确的实验室诊断的必要性2。婴儿的水疱通常偏向于四肢和尿布周围,但随着儿童的发育,水疱的模式通常会变得更具其亚型的特征。例如,在局灶性的EBS中,水疱主要形成在脚上,而在中度或重度DEB亚型中,皮肤脆性会在膝盖和肘部等骨隆处变得更加明显。虽然重度JEB的婴儿出生时皮肤起疱相对较少,但过几个月里,面部、耳朵和远端手指的特征性肉芽组织变得更加突出和独特。在KEB中,儿童早期水泡渐消退,光敏性和进行性皮肤异色病变得更加明显。,EB的一些后遗症是不可逆的和进行性的,例如DEB的皮肤和口腔粘膜瘢痕或指甲脱落,因此随着年龄的增长,这些后遗症往往会变得更加明显。EB的一些后遗症是不可逆的和进行性的,例如DEB的皮肤和口腔粘膜瘢痕或指甲脱落,因此随着年龄的增长,这些后遗症往往会变得更加明显。在严重的EBS中,婴儿会有非常严重和广泛的皮肤起泡,这一亚型可能会有致命的病程。然而,自然历史是一个随着时间的推移而逐渐改善的历史,因此成年人的水泡可能非常有限,主要局限于肢端部位。伴有斑驳色素沉着的EBS的临床特征也随着时间的推移而变化,通常伴随着整个童年期间水泡的改善,同时伴随着与先前的起泡和点状掌跖角化病无关的特征性色素变化的发展。带有KLHL24突变的中度EBS是值得注意的,因为它在出生时就有严重的皮肤脱落,这种情况会随着年龄的增长而改善,而且在成年早期就会发展成心肌病。同样,在带有PLEC突变的EBS中,SF在婴儿期和成年期之间的任何时间点都伴随着进行性肌营养不良的发作,也与心肌病有关。水疱的程度和模式可能在不同形式的EB中有所不同。例如,RDEB Inversa通常在生命早期包括中等程度的全身性水疱,但是在童年到成年的后期,易患部位变为皱褶部位。痒疹DEB也会随着时间的推移而演变,最初在小腿上会出现瘙痒样结节和线状病变,随着时间的推移,通常会扩散到更近端,也会扩散到手臂上。特定瘙痒特征的出现可能非常延迟,在成年后期开始出现83。类似地,局灶性胫前DEB的分布随着年龄的增长而变化。在晚发性JEB中,SF倾向于在儿童期中期开始,进行性硬皮病样萎缩,随后发生指甲改变。一些婴儿期重度JEB的病例与自发改善和长期存活有关;在这些病例中,LAMB3突变导致b3层粘连蛋白链被截断,但部分功能已被假定为导致中间临床症状47,84,85。不同亚型EB中不同分布模式及其随时间波动的机制尚不完全清楚,但可能在蛋白质水平上反映了特定的遗传后果。进一步阐明EB致病基因和其他遗传修饰物的基因型-表型相关性,可能会及时提供一些澄清。除了疾病特有的自然病史外,EB还可能伴随着许多继发性并发症,这些并发症随着时间的推移而发展,通常取决于EB类型的一般严重程度,以及环境和混杂因素,如细菌定植。例如,贫血、骨密度降低、肾损伤、进行性皮肤挛缩和鳞状细胞癌的发展都是严重RDEB的潜在并发症,但这些并发症是否或何时发生存在个体间的差异。EB分类的相关性和前景修订随着诊断和研究的发展,对于临床医生处理EB患者(用于咨询、预测、随访和筛查并发症)和研究人员来说应该是一个有用的工具。新出现的治疗选择和临床试验开辟了新的视角,并强调了分子遗传学和基因型-表型相关性对预测精确医学治疗选择的重要性。EB相关蛋白在确保细胞的机械稳定性和粘附性以及结构和功能上的特殊性(如层粘连蛋白332、整合素α6β487或胶原XVII88控制角质形成细胞的干性)具有独特的作用。然而,有一些共同的致病机制,如慢性组织损伤和炎症,适用于所有/几种类型的EB89。一些治疗原则,如诱导PTC突变的通读90-92,RNA治疗(例如,外显子93的反义寡核苷酸跳过93)或调节蛋白质错折叠94,在了解个别突变及其后果的前提下,可以应用于不同的基因/蛋白。因此,在分子缺陷的基础上对EB和SF疾病进行细分,并对精确药物的突变进行分层44是未来一个诱人的挑战。
窦丽敏医生的科普号 2020年02月12日3856
0
1
2020年02月12日3856
0
1
-
遗传性大疱性表皮松解症的临床表现与检查
一、概述 遗传性大疱性表皮松解症是一组罕见的单基因遗传性皮肤病,由编码皮肤结构蛋白的基因突变导致,呈常染色体显性或隐性遗传。主要表现为皮肤或黏膜脆性增加,受到轻微外伤或摩擦后出现破损、水疱或大疱改变。临床症状为水疱、大疱、皮肤色素异常、表皮缺失等。患儿皮肤像蝴蝶翅膀般脆弱,因此此病患儿又称蝴蝶宝贝。可引起手指并指、贫血等并发症,严重者可危及生命。并发症有手指并指、食管狭窄、牙釉质发育不全、贫血等。水疱等症状结合基因检测等检查可诊断。患者预后与疾病类型、基因突变位点及护理情况等有关。2018年5月11日,国家将遗传性大疱性表皮松解症被录入首批罕见病目录。 二、类型与临床表现 根据电镜下皮肤水疱在发生的显微结构的位置,可将此病分为单纯型、交界型、营养不良型及Kindler综合征。 1.单纯型遗传性大疱性表皮松解症 出生时或出生不久有水疱、大疱,多出现在手、肘、膝、足等易摩擦部位。属于常染色体显性遗传性疾病;致病基因有2个,KRT5,定位于12号染色体q13区域;KRT4,定位于17号染色体q12-q21区域。多数患儿在水疱愈合后还会有皮肤色素异常,肤色变浅或加深,但通常不会留下瘢痕或者皮肤萎缩。部分会出现口腔黏膜水疱或溃疡,但不太严重。甲营养不良、甲变厚、毛发稀疏等表现通常会缺如或程度较轻。 2.交界型遗传性大疱性表皮松解症 患儿症状大多较重。属于常染色体隐性遗传性疾病;突变基因有XVII型胶原基因与板层素基因突变所致。多数出生后即有大面积皮肤水疱、糜烂或表皮缺失。水疱愈合后可遗留瘢痕或皮肤萎缩。可以出现大片毛发脱失、甲脱落、牙釉质发育不良、吞咽困难、声音嘶哑、排尿困难及眼部翼状胬肉等。 3.营养不良型遗传性大疱性表皮松解症 营养不良型遗传性大疱性表皮松解症有常染色体显性遗传和常染色体隐性遗传两种遗传方式。呈常染色体显性遗传的症状较轻;常染色体隐性遗传的通常症状较重,皮肤癌变率高。Ⅶ型胶原基因(COL7A1)突变是致病基因。 患者大多在出生后出现小腿前面到足部皮肤缺失,肘、腹股沟等摩擦部位反复出现水疱、大疱。水疱愈合后,大多留有皮肤萎缩,看上去像羊皮纸。 患者有严重营养不良,发育迟缓,第二性征发育延迟,乏力等,易发生感染。此外,患者患皮肤癌的概率也相对增高。还会出现指、趾屈曲挛缩甚至并指;张嘴幅度变小,食管狭窄出现吞咽困难,便秘或肛门反复出血,眼部异常增生物等。 4.Kindler综合征 出生不久后,皮肤受到摩擦后出现水疱和大疱;随年龄增长,水疱及大疱逐渐减少,皮肤部分情况变好,但同时皮肤开始出现明显的光敏反应,可能有光暴露部位皮肤异色改变。患者水疱愈合后可能有皮肤萎缩、指纹缺失、轻度并指等表现。 三、病因 本病主要源于编码皮肤结构蛋白的基因突变,可呈常染色体显性或隐性遗传。目前本病已知有20个不同致病基因。 四、检查 1.免疫荧光检查 针对各个不同致病基因编码的结构蛋白进行免疫荧光染色,可以初步确定可疑致病基因。 2.组织病理学检查 皮肤组织病理学检查可见表皮内或表皮下水疱,真皮炎性细胞通常较少。通常组织病理性检查不特异。 3.透射电镜检查 透射电镜检查可确定皮肤水疱裂隙的层次,确定大的分型。 4.基因检测 基因检测发现致病基因的致病性突变位点,是准确诊断的前提。
窦肇华医生的科普号 2019年09月24日2653
0
0
2019年09月24日2653
0
0
-
大疱性表皮松解症的家庭护理
大疱性表皮松解症是一种遗传性疾病,由基因突变引起,部分为显性遗传,部分为阴性遗传,产检超声无法检出,往往在新生婴儿娩出当时或者前几天发现皮肤缺损或者全身皮肤多发水疱发现。近2年来已诊治大疱性表皮松解症
闫果林医生的科普号 2019年03月27日1914
1
2
2019年03月27日1914
1
2
-
遗传性大疱性表皮松解症治疗的新希望
这次在美国皮肤科医生年会(AAD)会议上,遇到了Phoenix Tissue Repair公司(一家专注于罕见病治疗方法和药物开发的公司)的王乐毅博士(副总),聊及到隐性遗传性营养不良型大疱性表皮松解
林志淼医生的科普号2019年03月04日8246
13
31
-
遗传性大疱性表皮松解症的日常护理
EB的治疗很重要,平时的护理也同样不能忽视 护理:减少摩擦及受力减少热刺激预防水疱感染避免继发损害营养支持:增加高蛋白物质及富纤维食物摄入,避免质硬食物鼓励定期排便适当日晒 功能锻炼:关节活动口腔、眼睑部位多学科护理 手外科矫正并指、粘连 消化科扩张狭窄食管耳鼻喉科减轻喉部狭窄,避免窒息 口腔科矫正张口受限、安装义齿愿大家能早日恢复健康。
林志淼医生的科普号2012年11月22日3215
1
2
-
遗传性大疱性表皮松解症的诊断流程
EB诊断流程:采集病史→电镜检查→免疫荧光→基因检测→回顾分析为何需要基因诊断? 基于基因突变位点的EB诊断有利于: 明确疾病的病因 准确判断疾病发展和预后 确定EB患者的护理方案 确定患者EB的遗传方式 为患者家庭提供产前诊断 为今后基因治疗奠定基础 更好认识EB的发病机制透射电镜免疫荧光 验证裂隙位置:IV型胶原抗体 确定缺陷蛋白:各种结构蛋白抗体基因诊断 PCR-DNA测序方法 诊断金标准 确定基于突变位点的最终分型基因诊断须注意事项 尽量提供详尽病史,特别是婚育史(近亲?) 血样采集尽可能全面,包括所有患者及家系相关健康人 采血每人需要2管,放置在EDTA抗凝紫色管,可常温保存2周,可进行快递 患者尽可能提供皮肤组织标本 基因检测需等待荧光和电镜结果,因此通常结果会比较慢(平均2个月) 基因检测存在无法得到结果的风险,甚至结果无法判定的可能性
林志淼医生的科普号2012年11月22日3245
2
0
相关科普号

王德进医生的科普号
王德进 副主任医师
杭州市第三人民医院
皮肤科
515粉丝8.5万阅读

刘波医生的科普号
刘波 无职称
北京积水潭医院
手外科
4578粉丝40.1万阅读

李明医生的科普号
李明 主任医师
复旦大学附属儿科医院
皮肤科
1.2万粉丝22万阅读
-
 推荐热度5.0杨宝琦 主任医师山东第一医科大学附属皮肤病医院 皮肤科
推荐热度5.0杨宝琦 主任医师山东第一医科大学附属皮肤病医院 皮肤科天疱疮 33票
皮肤病 5票
痤疮 2票
擅长:大疱性皮肤病(天疱疮、大疱性类天疱疮等)、重症银屑病、痤疮、湿疹、玫瑰糠疹、特应性皮炎、带状疱疹等皮肤病的临床诊疗。不包括性病和激光美容。 -
 推荐热度4.5王明悦 主任医师北京大学第一医院 皮肤性病科
推荐热度4.5王明悦 主任医师北京大学第一医院 皮肤性病科天疱疮 22票
药物性皮炎 1票
扁平苔藓 1票
擅长:常见皮肤病及性病。自身免疫性大疱病如天疱疮、类天疱疮等。银屑病。 -
 推荐热度4.5尚盼盼 主治医师北京大学第一医院 皮肤性病科
推荐热度4.5尚盼盼 主治医师北京大学第一医院 皮肤性病科天疱疮 21票
皮炎 6票
银屑病 4票
擅长:天疱疮、类天疱疮、疱病,银屑病(牛皮癣);皮肤科常见疾病,如皮炎,湿疹,痤疮,脚气,股癣等