




精选内容
-
线粒体病孩子如何改善营养
线粒体病是由于线粒体DNA或核DNA缺陷引起的遗传代谢病,临床表现包括发育落后、肌无力、智力低下、癫痫发作、中风样发作、心脏病、胃肠道疾病、肝肾疾病等,线粒体病尚无有效治疗方案,主要为多学科对症干预和管理,其中营养干预既可起到支持作用,也可对某些类型线粒体病发挥治疗作用。线粒体病孩子改善营养的目的是保证蛋白质、能量和各种营养素供给,改善机体功能及提高生活质量。蛋白质、脂肪和碳水化合物的摄入量参考国家膳食推荐量。合并呕吐、吞咽障碍、喂养困难或胃肠蠕动障碍,导致摄入减少,可采取肠内营养(经口或管饲)支持。特殊饮食及部分营养素可能具有调节代谢和信号通路、改善线粒体呼吸链功能的作用。常用的有抗氧化剂,如维生素C、维生素E、α-硫辛酸、辅酶Q10等;改善电子传递,如辅酶Q10、维生素K、琥珀酸盐等;减少毒性产物,如二氯乙酸、二甲基甘氨酸等;辅酶,包括肌酸、左旋肉碱、烟酰胺、硫胺素等;这些营养素均被认为有可能改善线粒体功能,常被联合使用,即“线粒体鸡尾酒”疗法。此外,精氨酸可改善线粒体活性及血管舒缩状态,防止卒中样发作。生酮饮食是一种高脂肪、低碳水化合物、合理蛋白质和其他营养素的饮食,除了控制癫痫发作,生酮饮食还可改善患者认知及提高肌肉力量,减少mtDNA异质性。生酮饮食可改善MELAS、Leigh病、LHON、KSS、线粒体肌病和进行性外眼肌麻痹、电子传输链缺陷等多种线粒体病。
陆相朋医生的科普号 2023年07月11日
2023年07月11日 105
105
 0
0
 1
1
-
医生你有推荐线粒体脑肌病的医院或人选吗

 王丽医生的科普号
王丽医生的科普号 2023年03月26日25
0
0
2023年03月26日25
0
0
-
线粒体病
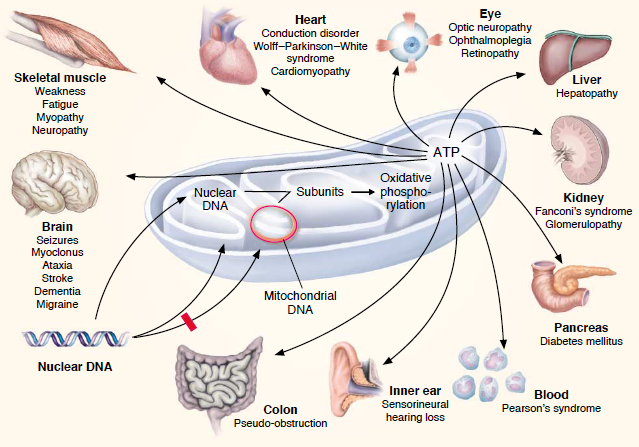
线粒体疾病是一组遗传代谢性疾病,当人体内的线粒体不能正常发挥功能时发生。线粒体是细胞内的细胞器,为细胞提供能量。它们含有自己的DNA,由母亲遗传。线粒体疾病可能影响各种器官和系统,如大脑、肌肉、心脏、肝脏、肾脏和眼睛。线粒体病可以引起各种症状,如生长迟缓、肌肉无力、视力和听力问题、学习障碍、癫痫、心脏问题和胃肠道障碍。线粒体疾病可能出生时就存在,也可能在更晚的年龄发生。有许多类型的线粒体疾病,根据它们的遗传原因或临床特征而有不同的名称。儿童中一些线粒体疾病的例子包括:KearnsSayre综合征:一种罕见的疾病,影响眼睛(导致眼睑下垂和眼动障碍)、心脏(导致心律失常)、肌肉(导致无力)和大脑(导致痴呆)。Leigh综合征:一种严重的疾病,影响大脑(导致逐渐失去智力和运动能力)、肌肉(导致无力)和其他器官(如心脏、肺和肾脏)。MELAS:线粒体脑肌病、乳酸酸中毒和类中风发作的缩写。它影响大脑(导致癫痫、头痛和中风)、肌肉(导致无力)和其他器官(如心脏)。MERRF:肌阵挛性癫痫和破碎红纤维的缩写。它影响大脑(导致癫痫)、肌肉(导致无力)和其他器官(如心脏)。MNGIE:线粒体神经胃肠病。它影响神经系统(导致麻木、疼痛和无力)和消化系统(导致腹泻、呕吐和体重下降)。Pearson综合征:一种罕见疾病,会影响骨髓(导致血细胞计数降低)和胰腺(导致糖尿病)。丙酮酸代谢障碍:一组影响细胞如何利用葡萄糖产生能量的疾病。会引起乳酸中毒(血液中乳酸积累)和各种神经系统功能障碍。Krebs循环障碍:一组影响细胞如何利用氧气产生能量的疾病。也会引起乳酸中毒和各种神经系统功能障碍。线粒体疾病可以通过基因测试或对血液或组织样本的生化测试进行诊断。治疗选择取决于疾病的类型和严重程度,但可能包括管理症状或预防并发症的药物,提供缺失维生素的膳食补充剂,改善肌肉力量的物理治疗,改善沟通的语言治疗,支持学习的特殊教育,替换受损器官的器官移植,或矫正缺陷基因的基因治疗。目前还没有治愈线粒体疾病的方法,但寻找新治疗方法的研究正在进行中。
 湘雅医院儿科科普号
湘雅医院儿科科普号 2023年03月02日350
0
1
2023年03月02日350
0
1
-
线粒体基因突变女性生育意愿调查问卷
您好,我们是安徽医科大学第一附属医院生殖中心线粒体遗传病研究团队,为调查线粒体遗传病患者的生育意愿与心理状况,我们设计了本问卷。本问卷为匿名填写,调查所得所有数据仅供研究使用,我们将严格保密,感谢您的支持与配合。
 安徽医科大学第一附属医院科普号2023年02月19日376
0
0
安徽医科大学第一附属医院科普号2023年02月19日376
0
0
-
线粒体病有哪些类型?
线粒体病是遗传缺陷性疾病。根据线粒体病变部位不同可分为:1.线粒体肌病。线粒体病变侵犯骨骼肌为主。多在20岁时起病,也有儿童及中年起病,男女均受累。2.线粒体脑肌。病变同时侵犯骨骼肌和中枢神经系统。多在儿童期起病,首发症状为眼睑下垂,缓慢进展为全部眼外肌瘫痪,眼球运动障碍,双侧眼外肌对称受累,复视不常见。3.线粒体脑病。病变侵犯中枢神经系统为主。包括Leber遗传性视神经病、亚急性坏死性脑脊髓病、Alpers病及Menkes病等。
曹玉红医生的科普号 2022年11月03日646
0
0
2022年11月03日646
0
0
-
什么是线粒体病?
线粒体病又称为线粒体细胞病,是遗传性氧化磷酸化功能缺陷致使ATP合成障碍、能量来源不足导致的一组疾病。线粒体基因或核基因突变均可致病。本病疾病种类较多,临床表现复杂多样,但以神经系统和肌肉病变为主。目前尚无有效治疗方法,以对症治疗、药物治疗、基因治疗等为主。
曹玉红医生的科普号2022年10月31日299
0
0
-
线粒体脑肌病并发高乳酸血症和卒中样发作综合征(MELAS)(头痛)
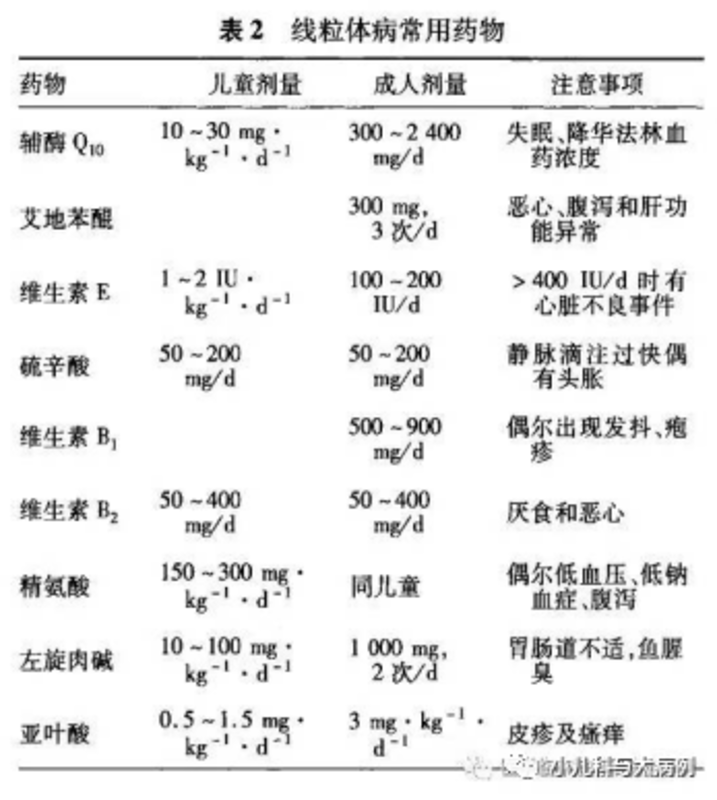
MELAS是线粒体脑肌病中较常见的一种类型,其临床主征是高乳酸血症、脑卒中样发作和癫痫。该病系线粒体遗传性疾病,没有治愈的方法,目前临床主要采用改善代谢的“鸡尾酒”疗法(见下图)以及对症支持治疗。 附病例小A,女,8岁,4天前出现发热,热峰39℃,没有其他不适,每天发热3~4次,服用退热药后体温可以下降至正常。2天后,到当地医院就诊,查血常规及CRP大致正常,给予“感冒药”治疗。本来以为就是感冒相关的头痛,可又过了1天,小A出现走路不稳,东倒西歪,伴有反应迟钝,表情呆滞,但没有抽搐、口角歪斜和肢体麻木。次日,出现头痛,以右侧额颞部为主,发热时疼痛明显,疼痛剧烈时伴有呕吐,热退后头痛缓解,不伴有头晕,偶有视物模糊,无重影。当地医生认为,小A感冒后出现走路不稳、表情呆滞和头痛,要警惕颅脑疾病,又因为有呕吐,因此同时做了头颅、腹部及盆腔CT扫描,但结果显示未见异常。此外,血常规、CRP、PCT、生化、尿液分析、腹部彩超、心电图等检查均未见异常。医生给小A体格检查发现,小A身高和体重都明显落后于同龄人,均低于-3SD水平。但是,家长说,小A早产,出生后呼吸困难,在新生儿科住院40天,期间还曾使用有创呼吸肌辅助通气。因此,家长说小A早产,从小抵抗力就不好,吃饭也比别人差一些,所以一直都瘦瘦小小的。医生问,以前曾有头痛发作吗?没有感冒的时候有没有头痛发作?小A父母说,孩子平时偶尔也有说头痛,半年前曾因“头痛和呕吐”就诊,期间也是做了头颅CT检查未见异常,住院治疗3天后好转。说当时没有感冒症状,只是单纯头痛。1个月前出现“近视”,看东西不清楚,测视力约近视250°。父母也觉得很奇怪,小A是个一年级的小孩,还没有读多少书,怎么就近视了,难道是因为经常给小A看手机导致的?也不对啊,父母从小担心小A眼睛受近视影响,有意控制看手机的时间。医生给小A体格检查发现,入院时小A身高及体重严重落后于同龄人-3个标准差,精神、反应差,营养不良貌,双眼对光反射稍迟钝,走路不稳,位置觉可疑异常(不能正确定位趾指),踝反射稍亢进,双侧巴氏征可疑阳性,脑膜刺激征阴性。头痛、视力改变、走路不稳以及神经系统体格检查异常,医生认为小A不是单纯感冒,很可能是神经系统出现问题了。小A头痛剧烈、持续,以头痛为切入点分析。小儿头痛的病因很多,一般看归纳为颅内疾病、颅外疾病和全身性病变。从上面的分析看,小A似乎是颅内疾病可能性大。导致头痛的颅内疾病包括:(1)感染性:如病毒脑、化脑、结核杆菌引起的结脑以及寄生虫引起的感染;(2)非感染性:如外伤、肿瘤、脑积水、良性颅内压增高或减低、脑血管畸形、自免脑、脱髓鞘病变、遗传代谢缺陷病和其他(如偏头痛等)。小A先有发热症状,医生自然要首先排除感染性病因。小A外周血血常规、CRP、PCT等感染指标不高,脑脊液检查正常(常规、生化、病原及涂片均未见异常),这些检查初步排除化脑和结脑的可能性。寄生虫脑炎也可以引起头痛剧烈,常见如广州管圆线虫感染,小A没有接触此类病原体的流行病学史,外周血及脑脊液的嗜酸性粒细胞也不高,因此也不支持。病毒引起的脑炎,脑脊液一般会有轻微改变,但也可以完全正常。小A脑脊液病毒病原学检查也未见异常,虽然不能完全排除病毒脑,但目前也没有支持的证据。此外,小A此前曾有多次在没有感冒的情况下出现头痛,特别是半年前那次,还因此住院数天。因此,医生考虑为非感染性颅脑疾病可能性大。没有外伤史,外院头颅CT未见异常,脑脊液压力正常,结合临床表现,因此初步排除肿瘤、脑积水、外伤和偏头痛等颅脑病变。脑脊液免疫学检查阴性,也初步排除自免脑。那么,就剩下两种可能性比较大的病因:颅脑血管畸形和遗传代谢缺陷病。医生给小A进一步做头颅MR平扫+增强、MRA检查,发现头颅血管未见异常,但是MR扫描发现:右侧顶颞枕叶大片皮层及皮层下白质改变。因此,考虑遗传代谢缺陷病导致的脑白质病变?儿童遗传代谢缺陷病相关脑白质病变的病因有很多,多数属于大分子病或重金属代谢异常所致,包括亚历山大病、肾上腺脑白质营养不良、异染性脑白质营养不良、球形性脑白质营养不良和佩梅病。但是小A不仅有脑白质的改变,病变还广泛累及皮层及皮层下组织,而且是单侧的,这一点与上述常见的脑白质病变所有不同。医生发现小A多次血气分析提示高乳酸血症:高乳酸血症(lacticacidemia,LA):乳酸生成过多或乳酸代谢障碍等引起血乳酸水平升高(>1.8mmol/L)。当乳酸>5mmol/L,伴代谢性酸中毒(血浆PH<7.25),称之为乳酸性酸中毒。高乳酸血症在儿童非常常见,多数假性高乳酸血症,即由于哭闹、止血带加压抽血、抗凝剂(用肝素-氟化钠较好)和送检时间过长有关系。这种情况,在小婴儿尤其常见。有些时候,甚至需要镇静下抽血,以减少由于哭闹引起的假性乳酸增高。小A已经8岁,抽血时很配合,没有哭闹,血气抽完及时检测,因此初步排除假性高乳酸血症。真性高乳酸血症分为原发性高乳酸血症和继发性高乳酸血症:原发性LA:由于丙酮酸进行氧化代谢或糖异生过程中的酶先天性缺陷所致。丙酮酸氧化代谢过程分3个阶段:丙酮酸氧化脱羧、三羧酸循环(TCA)、电子呼吸链(ETC)及氧化磷酸化作用。继发性LA:其他先天性疾病包括脂肪酸β氧化障碍、有机酸尿症、糖原累积病等所致。非先天性因素如窒息、感染、血管阻塞、肝病、药物等。那么,如何区分这些不同原因的先天性高乳酸血症呢?可以根据个人史、病史及辅助检查初步判定。此外,通过检测乳酸/丙酮酸,辅助判断。小A的乳酸/丙酮酸检查如下:图:根据不同的生化特点判别具体代谢疾病(该图来自广妇儿遗传与内分泌科,李秀珍的主任课件)图:高乳酸血症的诊治思路(注:该图来自广妇儿遗传与内分泌科,李秀珍主任课件)根据上述结果及诊断图表,小A外周血的乳酸和丙酮酸含量升高,乳酸/丙酮酸比值偏高,提示机体氧化压力偏高,考虑以下原因引起:线粒体能量代谢障碍(线粒体呼吸链异常或三羧酸循环酶异常)。基于以上推论,对小A进行线粒体脑肌病进行基因检测发现该患者为线粒体基因m.3243A>G突变,为已知致病的热点突变。由此,结合临床诊断:线粒体脑肌病并发高乳酸血症和卒中样发作综合征(MELAS)。许多疾病都可以出现头痛症状,甚至是感冒头痛也很常见。那么,如何识别线粒体脑肌病呢?包括以下征象:体格及大运动里程碑发育迟缓,身高及体重落后于同龄人,通常瘦小,大运动里程碑落后;运动不耐受,容易疲劳,懒动,乏力;运动和智力倒退,倒退比落后更有意义;非感染性头痛,伴有精神反应差、四肢肌力下降,甚至视力和听力下降或丧失;身体多毛,尤其是躯干和四肢多毛;不明原因的高乳酸血症,排除哭闹、止血带和送检的影响;头颅MR出现脑卒中样改变,可以是对称性,也可以是非对称性;头痛,但是排除常见的感染性疾病,影像学具有上述特点;家族曾有类似病史的病患,或者夭折。如果头痛儿童,出现以上征象的一条或多条,需要考虑线粒体脑肌病的可能性。
 付朝杰医生的科普号
付朝杰医生的科普号 2022年07月02日1555
1
0
2022年07月02日1555
1
0
-
线粒体脑肌病伴高乳酸血症和卒中样发作(MELAS)
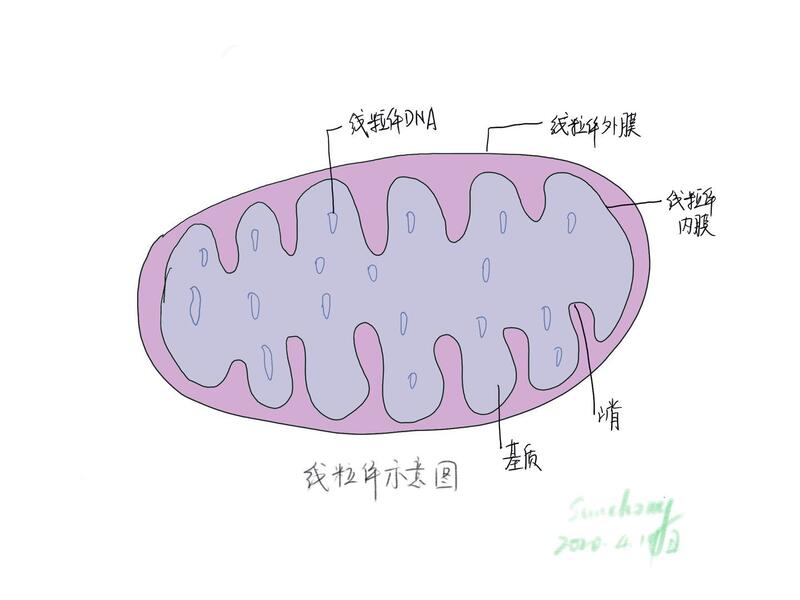
什么是线粒体脑肌病?线粒体是真核细胞的能量代谢中心,其内膜上富含呼吸链—氧化磷酸化的酶复合体,可以通过电子传递和氧化磷酸化生成ATP,为细胞提供进行各种生命活动所需要的能量。线粒体病就是一组由线粒体基因和/或核基因突变导致的线粒体呼吸链结构和功能异常的遗传性疾病,常累及多系统,其中神经及肌肉系统作为高耗能器官最容易受累,称为线粒体肌病/脑肌病。 MELAS的临床特点MELAS是线粒体病最常见的一种之一,它好发于20岁以前的年轻人,中老年仍有发病;以卒中样发作为特征性表现,具有母系遗传的特点。卒中样发作是MELAS特征性的表现,约发生在84.4%-99%左右的病人中,包括失语、偏盲、偏瘫、头痛、癫痫等;病灶所累及的区域并不按照血管分布且主要分布于皮层,故称为“卒中样”发作。每次卒中样发作可累及单侧或双侧大脑皮层,病灶可与前次发作部位相同或不同,症状持续数天到数周后恢复,但常遗留有脑萎缩。多次卒中样发作后往往会损害患者的运动能力、记忆力、智力、视力、语言能力等。其中癫痫发生在70.8%-96%左右的病人中,发作形式多样,部分发作和全面发作均有发生。反复发作的头痛在54-91%的病人中发生,常伴有呕吐,在卒中样发作的急性期头痛加重。听力损害发生在71-77%的病人中,常发生较早,多为感觉神经性耳聋,随着病程逐渐进展。虽然大多数患者早期生长发育正常,但是在55%-82%的患者中有身材矮小的症状。其他临床症状还包括发作性的脑病、记忆力减退、学习能力下降、肌强直、共济失调、眼外肌麻痹、视神经萎缩、视网膜色素变性、周围神经损害、精神障碍等。 其他系统中,心脏病变在18-30%的患者中可以被发现。心律异常、预激综合征、束支传导阻滞为心电图上常见的表现。部分患者心超可见左心室肥厚。胃肠道症状约发生在64-77%的患者中,食欲减退、恶心呕吐、便秘、腹泻、假性肠梗阻、胰腺炎均有发生。21-33%的患者伴有糖尿病,一型二型均可,一型多见。肾脏方面,范可尼综合征、肾炎性蛋白尿、局灶节段性肾小球硬化症也有报道。此外,患者还会有生长激素、甲状腺激素、性腺激素以及甲状旁腺素低下。 MELAS的实验室检查特点实验室检查中,血乳酸升高是线粒体脑肌病中常见的生化异常,与线粒体功能障碍无氧酵解增加有关。文献报道最高达94%的MELAS患者有血乳酸升高,但是需要注意的是判断静脉血乳酸升高时需排除由于止血带过紧或受试者过度活动造成的人为升高。乳酸升高的特异度在34%-62%之间,敏感度在83-100%之间。相较于血乳酸,脑脊液乳酸对于诊断线粒体病受人为影响小。血乳酸/丙酮酸比值对于判断线粒体电子传递链障碍更有意义,但是只适用于血乳酸升高的情况。肌酸激酶(CK)和乳酸脱氢酶(LDH)是另一组需要关注的生化指标,其中LDH更易升高且更具有提示意义。 MELAS的影像学特点实验室检查中,血乳酸升高是线粒体脑肌病中常见的生化异常,与线粒体功能障碍无氧酵解增加有关。文献报道最高达94%的MELAS患者有血乳酸升高,但是需要注意的是判断静脉血乳酸升高时需排除由于止血带过紧或受试者过度活动造成的人为升高。乳酸升高的特异度在34%-62%之间,敏感度在83-100%之间。相较于血乳酸,脑脊液乳酸对于诊断线粒体病受人为影响小。血乳酸/丙酮酸比值对于判断线粒体电子传递链障碍更有意义,但是只适用于血乳酸升高的情况。肌酸激酶(CK)和乳酸脱氢酶(LDH)是另一组需要关注的生化指标,其中LDH更易升高且更具有提示意义。 MELAS的肌肉病理和基因检测特点肌肉活检是MELAS确诊的重要依据。改良Gomori染色(MGT)见肌纤维周边线粒体异常沉积,表现为破碎红纤维(RRF) ,这些异常沉积的纤维在SDH染色中表现为周边蓝色深染,HE中也可见周边深染。另外,SDH强反应性血管(SSV)是较为特征性的表现。在COX中可见肌纤维活性增高,少量肌纤维活性降低或缺失。 基因检测是MELAS确诊的金标准。其中80%的MELAS与m.3243A>G突变有关,7.5%的MELAS与m.3271T>C有关,尚有10%MELAS基因突变不明。但需注意的是,并不是所有m.3243A>G突变均导致MELAS的发生,还其他表型或无症状的情况。 MELAS的治疗现有治疗方法主要针对改善线粒体氧化呼吸链功能、减少自由基损伤以及对症治疗,同时需要注意避免有害药物及刺激,合理饮食与锻炼及定期随访。现研究表明精氨酸对于卒中样发作的治疗和预防可能有效。卒中样发作的急性期静脉应用可以缓解除头痛和视野损害以外的所有卒中样症状,间歇期口服精氨酸则可以减少卒中样发作的频率和严重程度。此外辅酶Q10、艾地苯醌、肌酸、左卡尼汀、维生素B、 C、E、硫辛酸、糖皮质激素等也认为有益于MELAS患者,但均缺乏临床研究证据。对症治疗也十分必要。如抗癫痫治疗、止痛药的应用及营养支持。同时需要治疗并发症,包括心脏病、糖尿病、耳聋、精神障碍等。
 孙翀医生的科普号
孙翀医生的科普号 2020年04月19日7256
0
5
2020年04月19日7256
0
5
-
线粒体脑肌病伴乳酸血症和卒中样发作,北大医院就诊指南
1.会出现哪些临床表现?多数患者在发病前运动、智力发育基本正常,部分患者自幼运动不耐受。主要症状包括癫痫发作、脑卒中样发作(出现偏瘫、偏盲或皮层盲、失语、头痛、呕吐等表现)、痴呆、肌无力、耳聋等症状。该病还可累及其他系统,出现糖尿病、身体矮小、体毛增多、局灶性节段性肾小球硬化、心肌病或心脏传导异常、腹泻、假性肠梗阻等。2.什么是线粒体脑肌病伴乳酸血症和卒中样发作?线粒体脑肌病伴乳酸血症和卒中样发作(MELAS)是一种线粒体基因或核基因突变导致以脑病、脑卒中样发作、乳酸血症为主要症状的线粒体病,是线粒体病中最常见的类型之一。可以为母系遗传或散发,发病年龄从婴儿到成人期,以儿童期和青少年期发病居多。线粒体基因突变居多,且呈母系遗传规律,线粒体DNA 3243A>G突变是最常见的突变。核基因突变少见,遵循常染色体遗传。预后相对较差,多数患者在发病后10-15年死亡。3.怎样确诊?患者出现典型临床表现或头颅核磁改变,辅助检查发现血肌酸激酶和乳酸升高,需要考虑到该病。通过基因检查和肌肉活检就可以确诊该病。4.怎么治疗?补充维生素和各种辅酶,较常用的药物有辅酶Q10、维生素B1、维生素B2、维生素C、维生素E等。卒中样发作尽早使用L-精氨酸,可以减轻卒中样发作。患者反复出现癫痫发作,可选择合适的抗癫痫药物对症治疗。室性心动过速或肥厚性心肌病伴严重低血压患者需要安装植入式心脏复律除颤器等。5.找谁看病?可以到神经内科和儿科找专家看病,北京大学第一医院神经内科专家是王朝霞、俞萌、张巍、袁云,儿科专家是杨艳玲、熊晖和常杏芝。通过以下方式预约门诊:① “北京大学第一医院服务号” 微信公众号;②支付宝生活号关注“北京大学第一医院”;③网络预约请登录北京市预约挂号统一平台114网上预约挂号- 北京市预约挂号统一平台www.114yygh.com,实名注册后预约;④电话预约:(010)114。
袁云医生的科普号 2020年01月22日5481
0
12
2020年01月22日5481
0
12
-
线粒体脑肌病
线粒体是人体重要的生产能量的细胞器,是人体细胞的主要能量来源。线粒体的基本功能是氧化可利用的底物,通过呼吸链电子传递合成ATP。因此,线粒体的结构和功能异常往往导致整个能量代谢过程紊乱,从而产生一系列疾病,其中最常见累及神经系统的是线粒体脑病和线粒体肌病,这是一组与遗传、代谢有关的疾病。若不累及中枢神经系统而仅累及肌,称线粒体肌病;若合并有中枢神经系统损伤及症候的,称线粒体脑肌病。线粒体肌病是指因遗传基因的缺陷导致线粒体的结构和功能异常,导致细胞呼吸链及能量代谢障碍的一组多系统疾病;伴有中枢神经系统症状者,称线粒体脑肌病。此类线粒体疾病也可同时累及中枢神经系统,引起多种线粒体脑肌病。本病为一组临床综合征。线粒体脑肌病常见有下列临床综合征。一、线粒体肌病主要表现为以四肢近端为主的肌无力伴运动耐受不能,儿童和青年多见。病情进展缓慢,可有缓解复发。婴儿线粒体肌病有婴儿致死性和良性两种类型。致死性婴儿肌病多发生在出生后1周,表现为肌力、肌张力低下、呼吸困难、乳酸中毒和肾功能不全,多于1岁内死亡。良性婴儿肌病表现为婴儿期内肌力、肌张力低下和呼吸困难,1岁以后症状缓解,并逐渐恢复正常。最常见的基因异常为mtDNA3250位点上的突变。二、线粒体脑肌病伴高乳酸血症和卒中样发作(MELAS)线粒体遗传病方式方式(母亲遗传)。主要临床特征为小脑共济失调、肌阵挛或肌阵挛癫痫。40岁以前均可发病,10岁左右起病多见。母系亲属可呈现部分表现型,如仅有耳聋或癫痫。伴随症状可有身材矮小、精神运动发育迟缓、神经性耳聋、视神经萎缩、眼肌麻痹、颈部脂肪瘤、周围神经病、心脏病和糖尿病。主要的突变基因是MT-TL1,80%患者有该基因突变;其他的线粒体基因突变还有MT-TC、MT-TK等近20个基因突变。三、慢性进行性外眼肌麻痹临床表现为眼球运动障碍、眼睑下垂、短暂复视,多伴有易疲劳和肢体近端无力。20岁以前发病者多见,任何年龄均可发病。遗传方式可为家族性或散发性,部分为母性遗传,也可以是常染色体显性遗传。研究证实mtDNA有杂合缺失,也有mtDNA10909位点产生一个新的PvuⅡ酶切位点,由单个碱基置换。 四、Leigh病(亚急性坏死性脑脊髓病)为家族性或散发性线粒体脑肌病。于出生后6个月~2岁内发病。50%的患儿3岁前死亡。典型症状为喂食困难,共济失调,肌张力低下,精神运动性癫痫发作以及脑干损伤所致的眼睑下垂,眼肌麻痹,视力下降和耳聋。临床上见到幼儿出现反复发作的共济失调,肌张力降低,手足徐动及呕吐症状应考虑此病。部分为母性遗传,部分为常染色体隐性遗传或X连锁遗传。涉及本病的线粒体基因有MT-ATP6、MT-TL1及MT-ND1等近30种。五、Leber遗传性视神经病(LHON)LHON是一种线粒体遗传病。致病基因由母亲遗传给后代。该病外显率低,男性发病率比女性高4~5倍。任何年龄均可发病,通常为20~30岁。临床表现为急性或亚急性中央视野缺损。开始为单眼视物不清,数周或数月后双眼均受累,视力损害通常较重,可致全盲。还可有中枢神经的症状和体征、周围神经病和心脏传导阻滞。能导致LHON发病基因有MT-ND1、MT-ND2、MT-ND4、MTIND5及MT-ND6;此外,MT-CYB、MT-C3及MT-ATP6的突变,也能导致LHON,但一般都是散发病例。六、Wolfram综合征 主要临床表现是青少年发病的糖尿病和耳聋。此病具有发病年龄不定,程度不一,累及多器官和母系遗传特点。遗传基础为mtDNA中tRNA亮氨酸(1eu)基因的3243位点发生A→G碱基置换。此病患者与MELAS综合征的表型突变一致。
窦肇华医生的科普号 2019年08月20日3744
0
0
2019年08月20日3744
0
0
相关科普号

魏妍平医生的科普号
魏妍平 副主任医师
中国医学科学院北京协和医院
神经科
787粉丝21.8万阅读

邵玉凤医生的科普号
邵玉凤 主任医师
深圳市第二人民医院
神经内科
77粉丝33.7万阅读

吕平医生的科普号
吕平 主任医师
华中科技大学同济医学院附属协和医院
血管外科
243粉丝76万阅读
-

 推荐热度5.0蒋海山 副主任医师南方医科大学南方医院 神经内科
推荐热度5.0蒋海山 副主任医师南方医科大学南方医院 神经内科运动神经元病 18票
线粒体脑肌病 10票
重症肌无力 10票
擅长:运动神经元病(又称渐冻症、肌萎缩侧索硬化)、线粒体病、重症肌无力、先天性肌病、肌营养不良、格林巴利综合征、腓骨肌萎缩症、淀粉样变性周围神经病等肌病、周围神经病及神经系统疑难罕见病。可来就诊患者:肌无力、肌萎缩、肌跳、肌电图提示神经源性或者肌源性病变。 -
 推荐热度4.7林洁 副主任医师复旦大学附属华山医院 神经内科
推荐热度4.7林洁 副主任医师复旦大学附属华山医院 神经内科线粒体脑肌病 4票
重症肌无力 4票
周围神经病损 3票
擅长:周围神经病,线粒体病,肌病,重症肌无力,遗传性疾病基因报告解读及遗传咨询 -
 推荐热度4.2方方 主任医师北京儿童医院 神经内科
推荐热度4.2方方 主任医师北京儿童医院 神经内科小儿癫痫 4票
线粒体脑肌病 4票
抽动症 1票
擅长:小儿癫痫病、神经免疫性疾病、遗传代谢性疾病,线粒体肌病,药物难治性癫痫,婴儿痉挛症,