




精选内容
-
迟发型cblC型甲基丙二酸血症:临床、生化及基因特征
迟发型cblC型甲基丙二酸血症:临床、生化及基因特征【摘要】背景:cblC型甲基丙二酸血症是我国最常见的有机酸血症。迟发型cblC型甲基丙二酸血症(发病年龄>1岁)临床异质性明显,常易误诊。本研究旨在
 韩连书医生的科普号
韩连书医生的科普号 2024年03月13日
2024年03月13日 79
79
 0
0
 0
0
-
中国28例全羧化酶合成酶缺乏症患儿临床、生化和遗传分析
中国28例全羧化酶合成酶缺乏症患儿临床、生化和遗传分析凌诗颖、邱文娟、张惠文、梁黎黎、陆德云、陈婷、占霞、王瑜、顾学范、韩连书通讯作者:韩连书上海交通大学医学院附属新华医院、上海儿科研究所儿科内分泌与遗传代谢科,中国上海200092【摘要】目的:本研究旨在分析中国全羧化酶合成酶(HLCS)缺乏症患者的临床、生化和遗传学特征,并研究突变谱及其与表型的潜在相关性。方法:回顾性分析2006年至2021年28名HLCS缺乏症患者临床和实验室检测结果。结果:在28名患者中,有6名进行新生儿筛查,其中一人漏诊。因此,共23名患者因临床发病而被确诊。在所有患者中,24人出现了不同程度的临床症状,如皮疹、呕吐、抽搐和嗜睡,仅4例患儿至今仍无临床发病。患者血3-羟基异戊酰基肉碱(3-hydroxyisovalerylcarnitine,C5-OH)和尿丙酮酸、3-羟基丙酸、甲基枸橼酸、3-羟基异戊酸、3-甲基巴豆酰甘氨酸的浓度均大幅升高。在及时补充生物素后,患者的临床和生化症状均明显缓解,随访期间几乎所有患者的智力和身体均恢复正常。基因检测发现患者HLCS基因存在12个已知变异和6个新变异。其中,c.1522C>T变异最为常见。结论:我们的研究扩大了中国人群中HLCS缺乏症的表型和基因型范围,并表明及时进行生物素治疗,HLCS缺乏症患者的死亡率较低,预后较好。新生儿筛查对早期诊断、治疗和长期预后至关重要。关键词:全羧化酶合成酶缺乏症,HLCS,新生儿筛查,串联质谱1.引言:全羧化酶合成酶(HLCS)缺乏症(OMIM:253,270)是由于生物素代谢障碍引起的常染色体隐性遗传病,属于多种羧化酶缺乏症(MCD)的一种。据报道,全球HLCS缺乏症的发病率为1/200,000,中国发病率约为1/930,600。在日本,该病发病率较高,为1/100,000。HLCS催化生物素转化为羧化酶,包括丙酰辅酶A羧化酶(PCC)、3-甲基巴豆酰辅酶A羧化酶(MCC)、丙酮酸羧化酶(PC)和乙酰辅酶A羧化酶(ACC)。HLCS基因位于染色体21q22.1,长度约240kb,包含14个外显子。HLCS的缺陷会导致这些生物素依赖性羧化酶的活性降低,从而导致氨基酸、碳水化合物和脂肪酸的代谢失衡。HLCS缺乏症也被称为早发性MCD,因为大多数患者在新生儿期或婴儿早期临床发病,症状多样,包括皮肤损害、呕吐、肌张力低下、嗜睡、癫痫发作、代谢性酸中毒、高氨血症和发育迟缓。若不治疗,患儿会发展为严重的代谢性酸中毒,导致昏迷或死亡。早期诊断和及时生物素治疗可以预防神经系统后遗症和临床发病。因此,HLCS缺乏症符合纳入新生儿筛查计划的标准。目前,许多国家通过对干血斑上升高的C5-OH进行串联质谱检测来进行HLCS缺乏症的新生儿筛查。由于上述羧化酶活性降低,尿有机酸可升高,如乳酸、丙酮酸、3-羟基丙酸、甲基枸橼酸、3-羟基戊酸和3-甲基巴豆酰甘氨酸等。本研究分析了28名HLCS缺乏症患者的临床、生化和遗传特征,旨在总结目前中国HLCS缺乏症患者的基因变异谱、相应的临床表现和长期预后。2.方法:2.1患者:共纳入2006年至2021年上海新华医院儿科内分泌与遗传代谢科住院的28例HLCS缺乏症患者。收集其临床数据,包括发病时的临床症状和长期随访期间的症状HLCS缺乏症的诊断包括通过串联质谱法检测血C5-OH升高,气相色谱法检测尿乳酸、丙酮酸、3-羟基丙酸、甲基枸橼酸、3-羟基戊酸、3-甲基巴豆酰甘氨酸水平升高及基因检测HLCS变异。患儿父母或法定监护人签署了知情同意书,批准对其临床记录进行分析,并根据相关国家立法机构匿名公布其数据。该研究经新华医院伦理委员会批准(批准号XHEC-D-2022-063)。2.2代谢物检测:通过串联质谱法(MS/MS;AppliedBiosystems,API4000,加利福尼亚州,美国)检测干血斑上酰基肉碱水平,包括C5-OH、丙酰肉碱(C3)和乙酰肉碱(C2),同时计算C3/C2和C5-OH/C3的比率。通过气相色谱-质谱法(GC/MS;岛津制作所有限公司,QP2010,日本京都)测定尿有机酸的水平,包括丙酮酸、3-羟基丙酸、甲基枸橼酸、3-羟基戊酸和3-甲基巴豆酰甘氨酸。2.3遗传学检测:根据制造商的说明书使用DNA提取试剂盒(天根生物科技,北京,中国)从外周血中提取基因组DNA。对HLCS的14个编码外显子和侧翼区域均使用聚合酶链式反应(PCR)进行扩增,将PCR产物纯化后在ABI3700测序仪(AppliedBiosystems,福斯特城,加利福尼亚州,美国)上进行测序。利用ClinVar数据库、HGMD数据库以及既往文献来鉴定突变是否已被报道。新变异的致病性根据美国医学遗传学和基因组学学院(ACMG)标准和指南进行评估。对于新的错义变异,通过MutationTaster、PolyPhen-2、PROVEAN和SortingIntolerantFromTolerant(SIFT)预测潜在致病性,并通过phyloP和phastCons分析氨基酸保守性。2.4治疗:确诊后立即开始治疗,包括口服生物素和饮食调整。对于急性失代偿期的患者,初始治疗包括低蛋白饮食、补充生物素和左旋肉碱以及静脉注射葡萄糖和电解质液体疗法。再根据他们对生物素的反应和个人状况进行调整。终身使用生物素的推荐剂量为10-40毫克/天。2.5统计分析:未显着偏离正态分布的数据使用不配对t检验进行测试,非正态分布的数据使用Mann-WhitneyU检验进行测试。统计分析软件为GraphPadPrism5(GraphPadSoftwareInc.,圣地亚哥,加利福尼亚州,美国)。3.结果:3.1临床特征:本研究共纳入了28名确诊为HLCS缺陷的患者(17名男性,11名女性)。其中,6名患者接受了基于MS/MS的新生儿筛查,该检测以血C5OH作为生物标志物。然而,仅确诊了5例(5/28,17.9%)患者,其中1例在第一次筛查结果阳性后拒绝再次检测,在19天时临床发病确诊。余四名婴儿在生物素治疗后仍未发病。在这些患者中,仅1例患者(P24)因新生儿筛查漏诊,后出现面部、颈部、腋窝、躯干和尿布区域周围的鳞屑性红皮病、呼吸窘迫和乳酸性酸中毒等症状而被临床诊断。因此,大多数患者(23/28,82.1%)是由于具有典型的临床症状后通过MS/MS检测相关代谢物确诊。24名患者的发病年龄如表1所示。大多数患儿在1岁前出现代谢性酸中毒,发病年龄中位数为5月(7天-19月)。总体而言,2/3患者皮肤上出现不同程度的红斑疹。其中,仅一名患者(P27)主要表现为脸颊复发性湿疹等轻微皮肤症状,并在接受外用皮质醇和无乳制品饮食治疗后无任何改善。最终通过MS/MS以及GC/MS对其代谢物检测后诊断为HLCS缺乏症。再开始生物素治疗后,该患儿的皮损迅速恢复。HLCS缺乏症患者最常见的临床症状是皮损(66.7%,16/24)、呕吐(62.5%,15/24)、腹泻(37.5%,9/24)、呼吸衰竭(29.2%,7/24)、嗜睡(29.2%,7/24)、昏迷(20.8%,5/24)和癫痫(12.5%,3/24)。嗜睡、昏迷、癫痫发作等神经系统症状也是患者常见的临床表现,多发生于早发型患者,临床症状较严重。此外,三名患者(P1、P2和P5)均以癫痫发作为首发症状,生物素治疗效果显著,通常在给药后几分钟到几小时内症状缓解。共有5名患者在头颅MRI中出现异常,表现为大脑或皮质萎缩、白质减少或脑室扩大。3.2治疗前后代谢物检测:如表2所示,本研究中所有患者的血C5OH水平均远远超出正常范围,而血C3水平和C3/C2比率在近一半的患者中略有升高。此外,大部分患者C5OH/C3比率升高。同样,几乎所有患者尿有机酸水平异常,丙酮酸、3-羟基丙酸、甲基枸橼酸、3-羟基戊酸和3-甲基巴豆酰甘氨酸升高,表明HLCS缺乏。治疗后,除8例患者血C5OH水平、4例尿甲基枸橼酸、3-羟基戊酸超出正常范围外,余患者上述代谢物指标均明显下降。此外,所有患者治疗后代谢物水平较治疗前明显下降,差异有统计学意义(P<0.05)。3.3遗传学分析:对所有患者进行HLCS基因检测,结果在附加文件1:表S1中。共10名患者(35.7%)发现HLCS基因纯合突变。本研究共检测出17种不同突变,包括14种错义突变,占等位基因的89.3%(50/56);1个无义突变和2个缺失。其中,最常见的是c.1522C>T(p.R508W),占41.1%(23/56)。HLCS基因检测到6个新突变,包括c.663_664delCA、c.1825C>T(p.P609S)、c.1397G>T(p.G466V)、c.1433C>T(p.T478M)、c.2057T>G(p.I686S)和c.1481G>T(p.G494V)。五种新突变(P609S、G466V、T478M、I686S和G494V)的生物信息学特征如表3所示。3.4预后:在长达15年的随访中,患儿总体预后较好。就目前的健康状况而言,共有23例(82.1%)患儿发育正常,3例(10.7%)患儿死亡,2例(7.1%)患儿失访。三名患者死亡原因均是未及时诊断及生物素治疗而导致严重代谢性酸中毒,表现为呕吐、嗜睡和昏迷等。由于及时补充生物素,余23名患者均表现出代谢稳定性,神经、运动系统发育正常。4.结论:本研究对28名HLCS缺乏症患者的临床症状、生化特征、预后和遗传学结果进行分析。HLCS缺乏症患者主要临床表现是皮肤损害和代谢性酸中毒。若及时生物素治疗,能预防大部分患者临床发病,有利于良好预后。此外,本研究结果表明c.1522C>T是中国人群中最常见的突变,基于MS/MS的新生儿筛查有利于早期诊断及预后。
韩连书医生的科普号2024年03月13日12
0
0
-
新生儿筛查确诊538例cblC型甲基丙二酸血症患者预后分析
新生儿筛查确诊538例cblC型甲基丙二酸血症患者预后分析凌诗颖、毋盛楠、帅瑞雪、于玥、邱文娟、卫海燕、杨池菊、徐鹏、邹卉、封纪珍、牛婷婷、胡海利、张惠文、梁黎黎、陆德云、龚珠文、占霞、季文军、顾学范
韩连书医生的科普号2024年03月13日26
0
0
-
希特林蛋白缺乏症是什么病?
1.希特林蛋白缺乏症是一种遗传性代谢遗传病,也是一种继发性尿素循环障碍2.病人通常不喜欢含有大量碳水化合物的食物,如米饭、面条或蛋糕、面包。相反,他们喜欢含有脂肪和蛋白质的食物,如肉类、牛奶、乳制品、油炸食品和坚果。3.患者可以通过适当的饮食管理和医生的监测过正常的生活。4.由于这种疾病还存在着脂代谢异常,包括脂肪合成过多、脂肪酸氧化障碍,极有可能导致脂肪肝,甚至并发肝脏肿瘤。
贾钰华医生的科普号 2024年02月26日118
0
22
2024年02月26日118
0
22
-
希特林蛋白缺乏症-----一种爱吃零食的遗传病
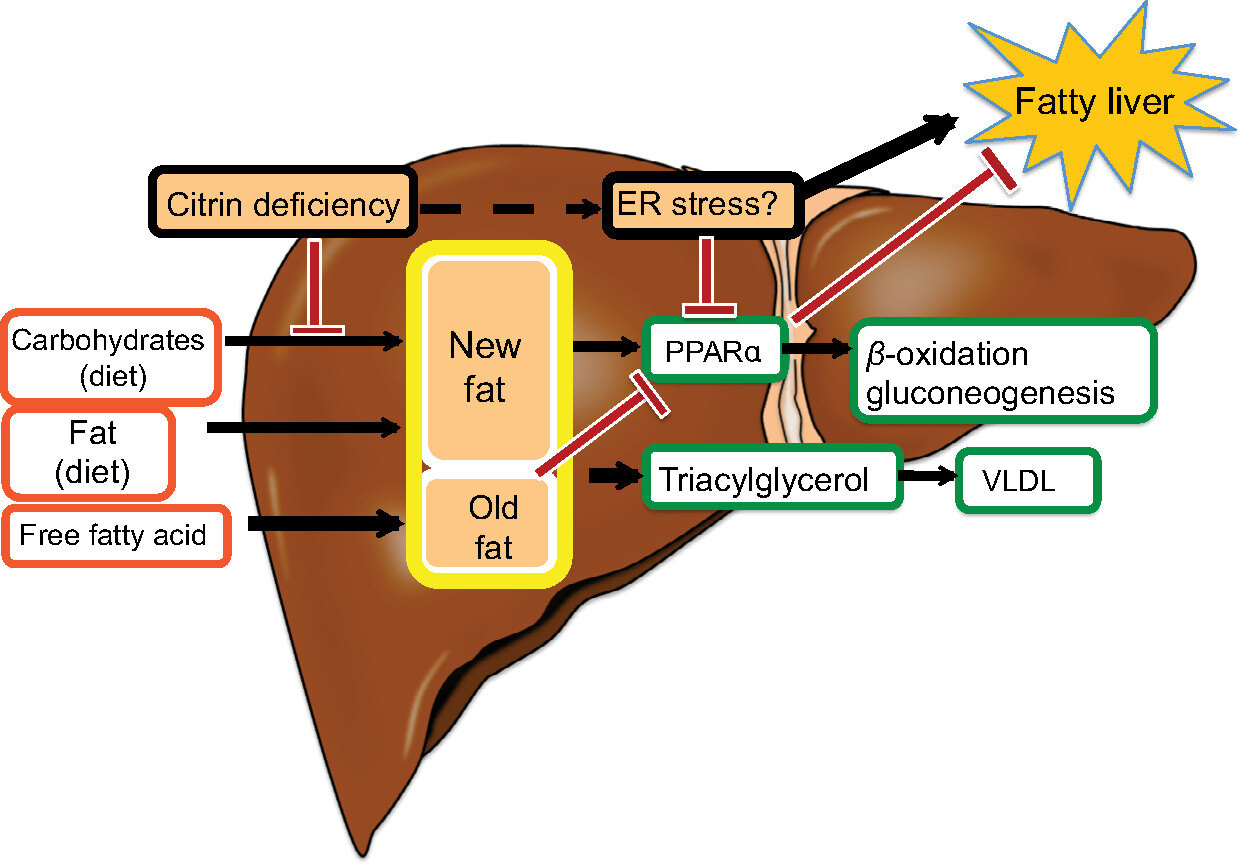
什么是希特林蛋白缺乏症?希特林蛋白缺乏症是一种遗传性代谢遗传病,也是一种继发性尿素循环障碍。病人通常不喜欢含有大量碳水化合物的食物,如米饭、面条或蛋糕、面包。相反,他们喜欢含有脂肪和蛋白质的食物,如肉类、牛奶、乳制品、油炸食品和坚果。患者通常可以通过适当的饮食管理和医生的监测过正常的生活。病因我们的身体里大约有3万个基因。每个基因产生不同的蛋白质,这些蛋白质构建了我们身体的蓝图。其中一个基因被命名为SLC25A13。这种基因产生一种叫做“希特林”的蛋白质。“希特林”蛋白的作用是帮助分解食物,为细胞产生能量,从而为我们的身体提供能量。希特林蛋白在细胞中起作用。在细胞内,希特林蛋白带着一种叫做谷氨酰胺的氨基酸从细胞质进入线粒体,在返回的途中,它从线粒体中获取另一种叫做天冬氨酸的氨基酸,并将其带到细胞质。在希特林蛋白缺乏时,SLC25A13基因发生了一些变异。SLC25A13基因突变要么不产生希特林蛋白,要么产生不完整或功能失调的希特林蛋白。因此,本应由希特林蛋白发挥的功能无法发挥,导致身体某些部位的代谢功能出现障碍,并诱发一些症状。据估计,在亚洲的一些地区,每40个人中就有1个人携带SLC25A13基因突变。需要2个变体才能成为患者,因此在亚洲一些地区,希特林蛋白缺乏症的发生频率假定为1/40x1/40x1/4=1/6400临床表现希特林蛋白缺乏引起的新生儿肝内胆汁淤积症(NICCD)发病时期:出生后到1岁常见症状:黄疸延长,肝内胆汁淤积,大便发白,体重停滞,半乳糖血症,瓜氨酸血症,低血糖。有些患者没有经历NICCD阶段。许多NICCD病例在1岁时自然消退,但一些严重病例可能需要治疗,包括肝移植。2.适应期/代偿期发病时期:1岁后或自NICCD恢复后常见症状:发育不良,低血糖,疲劳,腹痛,脂肪肝。请注意,每个患者可能会出现不同的症状,有些患者没有任何症状。从1岁开始,患者的食物偏好偏向高蛋白、高脂肪和低碳水化合物的饮食,这种食物偏好在大多数希特林蛋白缺乏症患者中都有体现。这种特殊的营养平衡在抑制症状方面起着非常重要的作用,是健康生活的关键。3.瓜氨酸血症II型(CTLN2)发病时期:主要发生在成年期,但也可以在青少年中见到常见症状:复发性高氨血症(血氨水平升高)神经精神症状:谵妄、攻击性、易怒、多动、定向障碍、躁动、嗜睡、记忆丧失、震颤、抽搐和昏迷在某些情况下,如果没有适当的强制治疗,患者可能会出现脑水肿,从而导致死亡大多数患者能够通过维持适当的饮食来避免CTLN2的发作希特林蛋白缺乏症的治疗目前,希特林蛋白缺乏症的主要治疗方法是饮食管理和MCT(中链脂肪)的摄入饮食管理保持低碳水化合物,高蛋白,高脂肪的饮食保持低碳水化合物、高蛋白和高脂肪的饮食是很重要的。当婴儿开始吃更多的固体食物,从食物而不是牛奶中获得更多的能量时,这种趋势就会变得明显。这是一个非常重要的功能,以“食物偏好”的形式补偿希特林蛋白缺乏的代谢问题。患者倾向于自然地保持他们喜欢吃的食物对他们来说舒适的PFC比例。换句话说,病人可以通过自己选择食物来保持健康。请注意不要摄入过多的碳水化合物。一次性摄入大量碳水化合物(糖)和长时间过量摄入碳水化合物(糖)可导致CTLN2发病。根据日本遗传代谢紊乱学会发布的2019年希特林蛋白缺乏指南,希特林蛋白缺乏患者的推荐蛋白质-脂肪-碳水化合物比例为蛋白质:15%-25%,脂肪:40%-50%,碳水化合物:30%-40%。尽量摄取多种脂肪来源,避免依赖动物脂肪,积极使用更健康的脂肪,如植物油等。强烈建议父母从儿童早期开始监督和教育他们的孩子,让他们意识到自己的状况,让他们自己管理饮食,帮助他们为未来的独立做好准备。建议患者通过自己准备食物或在青春期找到准备食物的方法来独立管理自己的饮食。由于代谢问题,希特林蛋白缺乏症患者的能量往往不足。对于任何年龄的患者来说,通过吃早餐、午餐、晚餐以及在两者之间吃零食来频繁地补充能量是非常重要的。MCT(中链脂肪)饮食为什么希特林蛋白缺乏症患者需要服用MCT饮食?希特林蛋白缺乏症患者不能消耗太多的碳水化合物,这就是为什么他们对碳水化合物含量高的食物产生天然的厌恶。相反,他们会吃很多高脂肪和高蛋白质的食物来获得能量,但普通的脂肪(如长链脂肪)可能会在以后增加健康问题。蛋白质产生能量的效率不高。MCT(中链脂肪)与普通脂肪的不同之处在于它直接向肝脏提供能量。这对希特林蛋白缺乏症患者很重要,因为他们的肝脏能量不足。由于MCT不常存在于食物中希特林蛋白缺乏症患者建议服用MCT油作为补充。MCT已被用于NICCD和CTLN2的治疗。此外,据报道,MCT油也对处于代偿期的希特林蛋白缺乏症患者有效。MCT饮食的推荐剂量为1克/公斤/天,如患者因胃部不适不能耐受该剂量,请相应调整剂量。关于如何摄入MCT的提示:1.NICCD患儿建议使用MCT浓缩配方奶粉/MCT补充母乳2.最好在用餐时服用MCT油,并在一天中分次服用3.建议食用MCT油的方法如下:(1)把它混合到饮料中(2)把它混合在沙拉里,或者加到煮熟的蔬菜里(3)把它加到汤里或者搅拌均匀蘸面包吃希特林蛋白缺乏症患者的建议忌酒即使少量的酒精也会对希特林蛋白缺乏症患者造成伤害,有时甚至可能危及生命。记住千万不要喝酒!定期检查婴儿患者建议每隔几个月就诊一次,饮食咨询一次;处于适应/代偿期的患者应每6-12个月就诊一次,饮食咨询一次。希特林蛋白缺乏症主要影响肝脏的健康,它可以在没有任何症状的情况下无声地恶化。定期的门诊检查对保持病情进展非常重要。不要输注高浓度葡萄糖和甘油在紧急情况下,输注高浓度葡萄糖或甘油治疗脑水肿可能会加重希特林蛋白缺乏症患者的病情。请告知医生,病人有希特林蛋白缺乏症。一般临时输注葡萄糖治疗低血糖和一般输注治疗腹泻或呕吐是没问题的。食欲恢复后,让患者吃高蛋白、高脂肪的食物,会恢复得更快。
 陈立医生的科普号
陈立医生的科普号 2023年11月22日171
0
3
2023年11月22日171
0
3
-
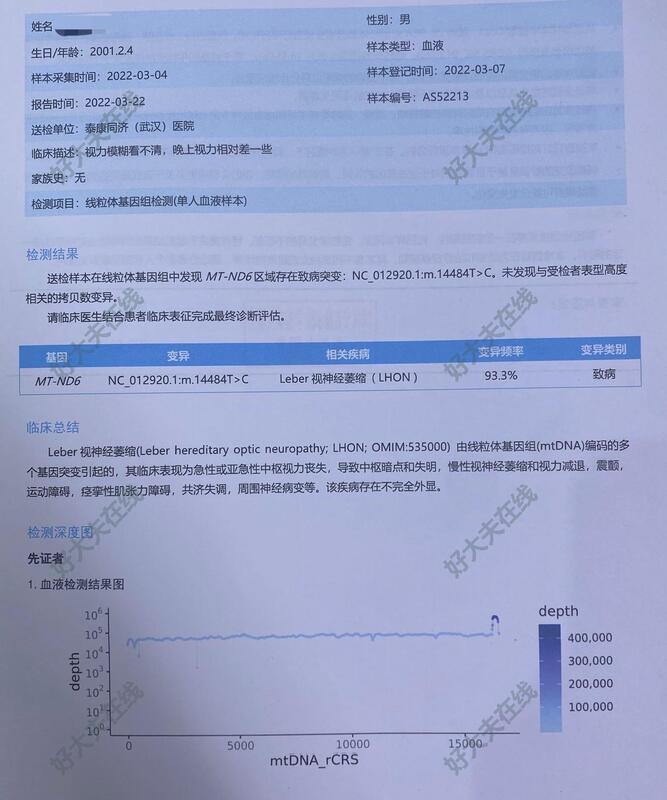
Leber遗传视神经
 陈中山医生的科普号
陈中山医生的科普号 2023年08月09日262
0
0
2023年08月09日262
0
0
-
早期容易被误诊为类风湿的法布雷病是如何诊断的,需要做什么检查?

 袁云医生的科普号
袁云医生的科普号 2023年03月24日167
0
5
2023年03月24日167
0
5
-
肉碱棕榈酰转移酶1缺乏症
一、概述肉碱棕榈酰转移酶1(CPT1)缺乏症是一组罕见的代谢障碍性疾病,是肉碱和线粒体脂肪酸代谢障碍疾病之一,是常染色体隐性遗传性代谢病。肉毒碱棕榈酰转移酶与乙酰辅酶A一起在线粒体膜内、外起转运乙酰基作用。由于线粒体膜外侧的肉碱棕榈酰转移酶1缺乏,肉毒碱作为载体将长链脂肪酸向线粒体内转运的机制出现障碍,中、长链酰基CoA不能顺利进人线粒体内进行β氧化,导致乙酰CoA生成减少,同时影响肝的生酮作用,长链酰基CoA等大量堆积,导致人体骨骼肌无法利用长链脂肪酸,可能引起血液中脂肪累积和肌痉挛,还可出现肌坏死、肌蛋白尿症、贮脂性肌病、低糖血症、脂肪肝和伴肌疼痛、疲倦及意识模糊的高氨血症等并发症。当葡萄糖摄人不足或其他疾病导致能量需求增高时,可出现肝损害及大脑功能障碍。一旦确诊,要尽早治疗,是预防及处理并发症的关键;补充肉毒碱,可改善预后。本病预后不佳;特别是并发症较多,且易死于严重并发症。二、临床表通常在禁食或疾病后发生,多于婴儿期或幼儿期发病。肉碱棕榈酰转移酶1缺乏症患者临床表现多样,分为迟发型、婴儿型、致死性新生儿型及急性脑病型。大多在出生后数小时至30个月发病。诱发因素常为饥饿、感染、腹泻等,起病急骤,类似Reve综合征发作,常复发,死亡率较高。脑部远期预后取决于低血糖的严重程度。新生儿型病情严重,患儿于出生数小时至数天内发病,低体温、呼吸窘迫、惊厥、喂养困难、昏迷、肝大、肝功能衰竭、心脏扩大,死亡率很高。迟发型患者常在儿童期发病,男性多见。过度运动、禁食和感染是常见的诱发因素,寒冷、睡眠不足、药物及全身麻醉可诱导发病。主要表现为肌痛、肌红蛋白尿、肌无力、肌强直及横纹肌溶解,严重者可引起肾衰竭、心肌病,甚至死亡。三、致病基因现已发现肉碱棕榈酰转移酶1有3种同工酶:肝型(CPTIA)、肌型(CPT1B)和脑型(CPT1C),均有组织特异性。1.肝型CPTIA除在肝中含量丰富外,还在肾、成纤维细胞及胰岛中表达,在心脏中也有表达。编码肉碱棕榈酰转移酶1的基因是CPTIA,定位于染色体11q13.3。CPTIA突变导致了肉碱棕榈酰转移酶1活性缺乏。2.肌型CPT1B主要表达于骨骼肌、心脏及棕色脂肪等组织。CPTIC仅在大脑中表达。CPT1A和CPT1B位干线粒体外膜上,催化长链酰基CoA与肉碱合成酰基肉碱。3.脑型CPT1C位于神经元内质网,不参与脂肪酸氧化代谢,可能与摄食行为和整体内稳态的调节有关。四、实验室检查1.血氨基酸及肉碱谱分析游离肉碱(CO)水平显著增高(>100μmol/L),多种中、长链酰基肉碱水平升高,尤其是棕榈酰肉碱(C16)、十八碳酰肉碱(C18)和十八碳烯酰肉碱(C18:1)、CO/(C16+C18)升高。2.常规检验急性期可见低酮性低血糖、代谢性酸中毒、血清肌酶增高、高血氨、转氨酶升高、高血脂、肝性脑病。某些患儿伴肾小管性酸中毒。3.基因检查CPTIA基因检出纯合或复合杂合变异,有确诊价值。五、诊断标准1.无特异性症状临床表现患者临床缺乏特异性症状与体征,临床诊断困难,死亡率高,对疑似患者应及早检查。2.血游离肉碱(CO)显著增高CO/(C16+C18)升高是必要条件。3.CPTIA基因检测阳性者,有助于确诊。六、治疗与预后1.基本原则避免饥饿,低脂高碳水化合物饮食,以减少低血糖的发生、减少脂肪动员的供能途径并增加糖原储备。2.左卡尼汀原发性肉碱缺乏症患者仅需补充左卡尼汀,疗效良好。3.禁用或慎用药物大环内酯类抗生素、丙戊酸钠、水杨酸类药物具有潜在的肝毒性,可能诱发Reye综合征,避免使用。4.女性患者或携带者女性患者或携带者孕期时容易发生急性脂肪肝、HELLP综合征;避免饥饿,坚持低脂高碳水化合物饮食,避免低血糖的风险,保护胎儿。5.新生儿筛查可以检出肉碱棕榈酰转移酶1缺乏症患者,在无症状时期或疾病早期开始治疗,避免器官损害,显著改善预后。七、遗传咨询与产前诊断为常染色体隐性遗传病,按照常染色体隐性模式咨询。1.患者父母致病基因携带者,每次生育时胎儿有25%的概率为患者,50%的概率为无症状携带者,25%的概率不携带父母来源的致病变异。2.生育过原发性肉碱缺乏症患者的夫妇再次妊娠前要做遗传风险评估和咨询。3.患者的健康同胞应进行基因分析及血游离肉碱和酰基肉碱谱分析,如为携带者,则需对配偶进行基因检测。4.基因诊断明确的家系在母亲再次妊娠11~13周采取绒毛或在16~22周抽取羊水,通过基因检查进行胎儿产前诊断。也可以选择植入前遗传学检测,规避患儿出生。5.携带者携带者常有轻度肉碱缺乏,需要在生育前、妊娠期、哺乳期补充左卡尼汀。6.孕妇患者原发性肉碱缺乏症患者怀孕后,仅需补充左卡尼汀,疗效良好。有人主张不建议进行产前诊断及不建议医学引产。
窦肇华医生的科普号 2023年03月10日561
0
0
2023年03月10日561
0
0
-
儿童反复出现手脚痛,少汗无汗,不早诊早治,可能让他少活30年_警惕法布雷病!指尖血检测就能排查
 张振洪医生的科普号
张振洪医生的科普号 2023年02月22日61
0
4
2023年02月22日61
0
4
-
伴脊髓与脑干受累及脑白质乳酸升高的脑白质病(LBSL )
一、概述LBSL为DARS2基因突变导致的常染色体隐性遗传病,主要表现为缓慢进行性小脑共济失调、痉挛和脊柱功能障碍。儿童或青少年起病,出现运动障碍、认知下降和感觉神经病。磁共振检查为不均匀、片状的脑白质受累,选择性累及包括脊髓在内的锥体束走形区域、包括内侧丘系等在内的感觉神经传导通路,小脑白质常受累,核磁共振波谱成像在病变区域可见倒置的乳酸峰,血﹣脑脊液中乳酸轻度升高。二、临床表现LBSL的疾病谱范围广,新生儿发病并在2岁前死亡,儿童期发病以及只有轻度损伤,成人发病类型缓慢进展。最常见的是儿童期发病,为缓慢进行性小脑共济失调、痉挛和背柱功能障碍。大多数患儿最初发育是正常的,运动功能障碍通常出现于儿童或青少年时期,偶尔出现在婴儿期或成人期。运动功能障碍包括小脑性共济失调、痉挛和背柱功能障碍,下肢较上肢易受累及,腱反射保留;下肢位置觉和振动觉减退,黑暗中行走困难;手指运动的灵活性受到不同程度的损害。部分患者有轴突神经病变,导致腱反射减弱或消失、远端肢体无力和感觉障碍。构音障碍随着时间逐渐进展。部分早期发病的患者有学习障碍,多数患者智力正常;可能发生轻微的认知能力下降。部分患者在轻微头部外伤后可出现意识水平下降、神经功能恶化和发烧。三、发病机制LBSL可归结为线粒体功能障碍。不同的线粒体氨基酸酰基-tRNA合成酶缺陷,通过影响相应的线粒体蛋白翻译,可导致多种线粒体疾病。LBSL是线粒体天冬氨酸t-RNA合成酶(mt-AspRS)缺陷。病致病基因DARS2位于1q25.1。绝大多数LSBL患者是DARS2复合杂合突变,也有纯合子错义突变。已经发现有60多种不同DARS2突变。线粒体分布于机体的各个细胞中,神经细胞较非神经细胞更易受第3外显子缺失的影响,神经细胞较非神经细胞对正常的含有第3外显子的mt-AspRSmRNA翻译效率也低,两种原因共同作用导致神经系统的易损性。电镜显示受影响的白质出现空泡改变和髓磷脂分裂。四、诊断标准1.影像学表现 LBSL诊断主要标准。(1)大脑白质非均质性点状或除U型纤维外均质成片的信号异常。(2)全段脊髓背柱及双侧皮质脊髓束信号异常,颈段脊髓核磁片即可显示出此异常。(3)延髓锥体束信号异常。次要标准:位于胼胝体压部、内囊后肢、延髓内侧丘系、小脑上脚、小脑下脚、三叉神经、中脑三叉神经束、延髓脊髓小脑前束、小脑白质的信号异常。LBSL患者磁共振波谱MRS检测异常脑白质可见乳酸峰增高,但并非所有患者都伴有异常白质的乳酸增高。2.基因检测 对符合MRI诊断标准的高度可疑病例,进行DARS2检测是最终诊断。绝大多数LBSL患者是DARS2复合杂合突变。DARS2有60多种不同基因突变。在LBSL的诊断过程中,要注意与多发性硬化(MS)、脊髓小脑性共济失调(SCA)、脊髓亚急性联合变性(SCD)、髓鞘化低下伴脑干脊髓受累及下肢痉挛的白质脑病(HBSL)及线粒体脑病(ME)做鉴别诊断。五、治疗关于LBSL的治疗,主要是支持性治疗,包括物理治疗和康复治疗,以改善运动功能,预防继发性并发症,如挛缩和脊柱侧凸等。以及对症治疗,如抗癫痫药物、特殊教育、语言治疗。六、预后LBSL的预后相对较好,除新生儿发病的严重病例外,该病病程呈现缓慢进展的过程。大多数儿童期发病的患者在十几岁、二十几岁或更晚的时候会部分或完全依赖轮椅,而成人发病的患者则一般不会依赖轮椅。
窦肇华医生的科普号2022年12月08日281
0
1
遗传代谢病相关科普号
贾钰华医生的科普号
贾钰华 主任医师
南方医科大学南方医院
中医内科
1万粉丝886.3万阅读

吴龙医生的科普号
吴龙 主治医师
首都医科大学附属北京朝阳医院石景山院区
妇产科
9867粉丝1399.7万阅读
陈中山医生的科普号
陈中山 主任医师
医生集团-湖北
线上诊疗科
827粉丝15.1万阅读
-
 推荐热度5.0万平 主治医师上海交通大学医学院附属仁济医院(东院) 肝脏外科
推荐热度5.0万平 主治医师上海交通大学医学院附属仁济医院(东院) 肝脏外科肝移植 82票
肝母细胞瘤 13票
遗传代谢病 8票
擅长:小儿肝病及罕见病的临床诊治,尤其是儿童遗传代谢病、胆道闭锁、胆汁淤积症、肝母细胞瘤等疾病的肝移植手术治疗,包括酪氨酸血症、尿素循环障碍(如鸟氨酸氨甲酰转移酶缺陷、HHH综合症、瓜氨酸血症、氨甲酰磷酸合成酶缺陷、精氨酸血症、精氨酰琥珀酸合成酶缺陷等)、甲基丙二酸血症、丙酸血症、糖原累积症、原发性高草酸尿症、戈谢病、尼曼匹克病、家族性高胆固醇血症、Caroli病、枫糖尿病、肝豆状核变性、进行性家族性肝内胆汁淤积症、Crigler-Najjar综合症、线粒体病等。 -
推荐热度4.5杨艳玲 主任医师北京大学第一医院 小儿神经内科
遗传代谢病 3票
肌病 1票
发育迟缓 1票
擅长:遗传代谢性疾病的诊断与治疗 -
 推荐热度4.5商晓红 主任医师山东省立医院 小儿内分泌科
推荐热度4.5商晓红 主任医师山东省立医院 小儿内分泌科遗传代谢病 5票
佝偻病 3票
小儿糖尿病 3票
擅长:儿童矮小症、性早熟、糖尿病、先天性肾上腺皮质增生症、各类佝偻病等内分泌疾病;甲基丙二酸血症、戊二酸血症、枫糖尿病、异戊酸血症、线粒体病、高胰岛素血症、低血糖、低血钾等遗传代谢病的防治有研究。